
产品优势
超长读长,直接获得的全长转录本序列。
Iso-Seq平均长度为2Kb左右,远大于一般转录本中典型基因的长度,可以轻松跨越从5’末端到3’-Poly A tail的完整转录本,无须拼接,直接测得转录本全长。
准确识别可变剪接事件,精准重构转录本。
以往基于二代短读长测序的转录组研究策略,通过reads拼接推测转录本结构,面对复杂的可变剪接,会产生许多错误拼接。采用Iso-Seq全长转录组,能够精准重构转录本结构。
可进行融合基因鉴定、可变聚腺苷酸化(APA)分析。
方案策略
| RNA样本量 | 建库策略 | 测序策略 |
| 总量 ≥ 1μg, 浓度 ≥300ng/μL | 反转录得到全长CDNA构建插入片段0.5-6Kb的cDNA SMRTbell文库 | 推荐数据量≥20Gb |
分析内容
| 有参转录组 | 无参转录组 |
| 1、全长转录本鉴定 2、与参考基因组比对 3、差异表达分析 4、富集分析 5、可变剪切分析 6、基因融合分析 7、Novel基因预测 8、APA分析 9、变异检测 10、IncRNA预测 | 1、全长转录本鉴定 2、构建 unigene集 3、功能注释 4、富集分析 5、SSR分析 6、CDS 预测 7、IncRNA 预测 |
结果展示
全长转录本鉴定
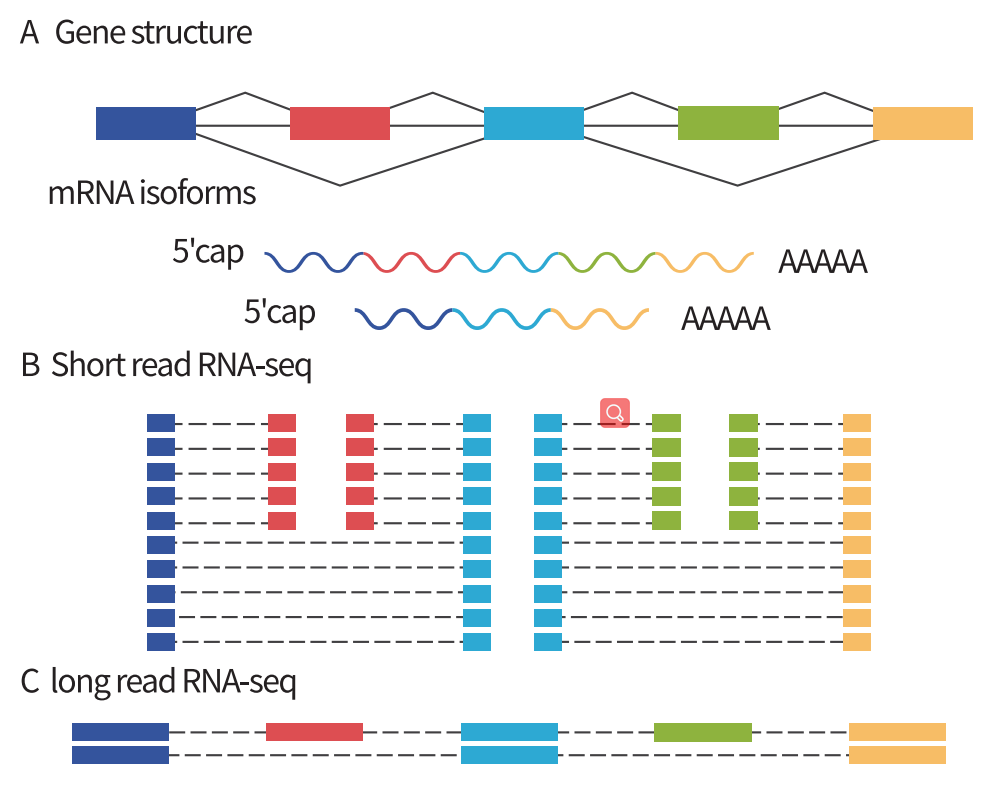
理论上,完整的插入片段序列(Reads of insert),一端为5’ primer, 另外一端为3’ primer,且 3’ primer前也存在polyA序列。因此通过判断5’primer, 3’primer, polyA的存在以及位置关系,将插入片段序列进行分类,同时包含5’primer, 3’primer, polyA并且相对位置正确的插入片段为完整插入片段序列,即Full-length reads。
 全长转录本鉴定流程
全长转录本鉴定流程
融合基因鉴定
融合基因(fusion genes)是指两个基因的全部或一部分的序列相互融合构成的嵌合基因。其可能是染色体易位、中间缺失或染色体倒置形成的。
 融合基因圈图
融合基因圈图
与参考序列注释比较
将去冗余后的转录本与参考注释中的转录本进行比较,然后提取比较结果信息,并将三代注释结果和参考注释结果进行合并。
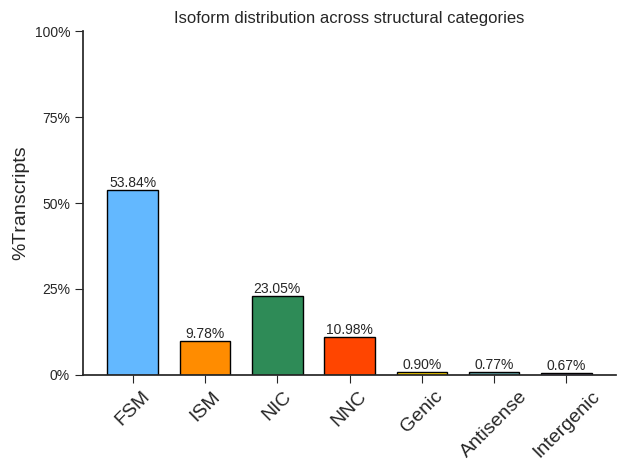 不同转录异构体分布
不同转录异构体分布
可变剪接分析
大多数真核基因转录产生的mRNA前体是按一种方式剪接产生出一种成熟mRNA分子,因而只翻译成一种蛋白质。但有些基因的一个mRNA前体通过不同的剪接方式(选择不同的剪接位点)产生不同的mRNA剪接异构体,这一过程称为可变剪接(或选择性剪接,Alternative Splicing),它是调节基因表达和产生蛋白质组多样性的重要机制。
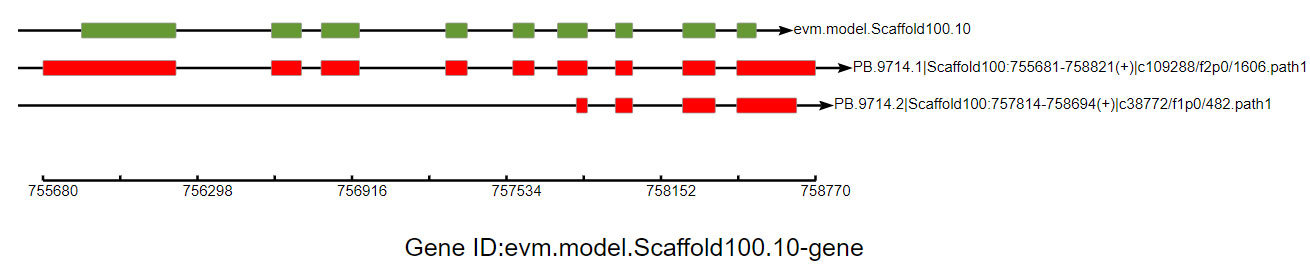 可变剪接事件基因结构
可变剪接事件基因结构
可变多聚腺苷酸化分析(APA)
可变多聚腺苷酸化(Alternative Polyadenylation, APA)是一种普遍存在于真核生物中的基因调控机制,是指一个转录本可以具有不同的多聚腺苷酸化位点,使不同的转录本具有不同的编码序列,或具有不同长度的3’非翻译区(3’UTR),从而使一个基因能够产生多种mRNA转录本,影响mRNA的功能、稳定性、定位和翻译效率。
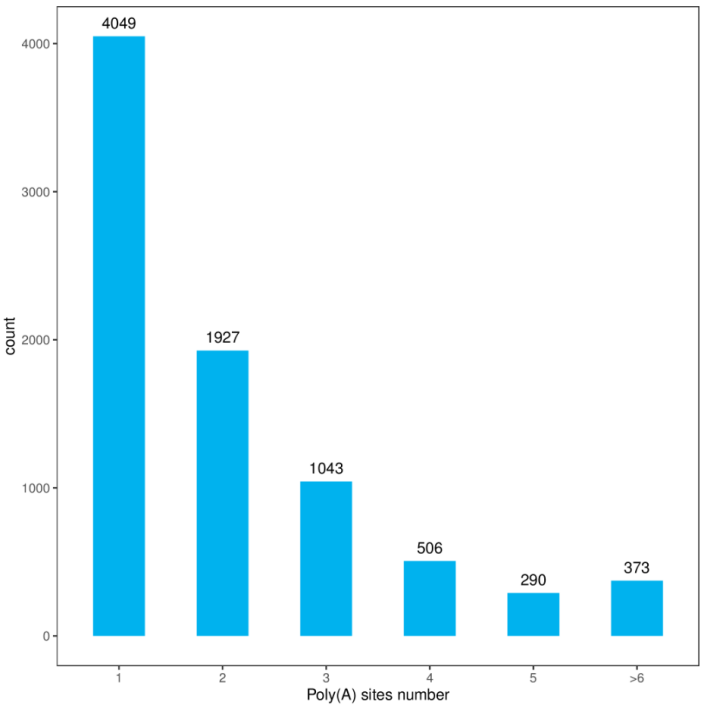 APA位点统计
APA位点统计
常见问题
1. Iso-seq全长转录组的优势是什么?
无需打断拼接,无需组装,可直接获得包含5’,3’UTR,poly A tail的全长转录本,因此可准确分析有参考基因组物种的可变剪切,融合基因,变多聚腺苷酸化等信息。
2. 是否需要构建不同片段大小的混合文库?
混合建库是针对之前的RSII平台,Sequel平台升级以后,在片段偏好性上有很大的提升,一般而言,构建0.5-6kb的文库即可得到和真实转录本分布一致的结果,不需要构建不同片段大小的混合文库。
案例解析
案例分析
案例解析
PacBio长读长测序揭示玉米转录组的复杂性
研究背景
玉米(Zea mays)是全球重要的农作物,也是研究植物转录组代谢通路的遗传模型。玉米基因组序列于2009年公布,后续利用EST和RNA-Seq转录组数据对其基因注释进行了补充。然而RNA-Seq中,短读长无法提供转录本全长序列,限制了可变剪接形式的界定,同时,短读长拼接会得到低质量的转录本,导致错误的基因注释。
冷泉港实验室等单位利用PacBio长读长测序技术,对玉米6个组织进行了全长转录组测序分析,在已有的玉米B73 RefGen_v3上发现了大量新信息,揭示了玉米基因表达的复杂性,研究成果发表在Nature子刊。
研究方法
- 取玉米自交系B73不同发育阶段的6个组织(根、花粉、胚芽、胚乳、幼雌穗、幼雄穗),提取mRNA, 反转录过程中加组织特异性barcodes,按照等摩尔比值cDNA平均混合,构建6种插入片段文库(<1, 1–2, 2–3, 3–5, 4–6 和>5 kb),不同的文库加上序列特异性的barcode,上机46个SMRT Cell进行全长转录组测序。
- 对6个组织进行二代RNA-Seq测序,每个样品三个重复。
- 通过已发表的甲基化数据,对isoform,IncRNA和non-IncRNA进行甲基化分析。
研究结果
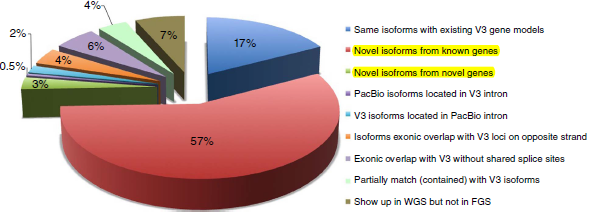
- 下机得到了3,716,604 reads,过滤得到接近一半的全长转录本序列(1,553,692,42%)。再处理得到643,330个高质量的转录本序列,其中606,145个序列(2%)能够比对到玉米RefGen_v3参考基因组上。 经聚类得到了111,151个isoform,对应26943个基因,涵盖了玉米RefGen_v3基因注释的70%。其中57% isoform来自已知基因位点的新isoform;2,803 (3%) 新isoform来自2,253 新位点。
- 已有研究中有1704个高度可信的LncRNA(平均长度为463bp)。本次分析得到了878个LncRNA,其中11个是已有研究确定的, 另外867个是新发现的LncRNA(平均读长为1kb)。
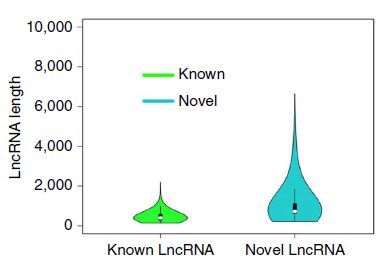
新发现和已确定lncRNAs长度对比
- 将PacBio isoforms与 Illumina短读长组装的isoform分析结果进行了比较,其中短读长数据运用两种分析方法(Cufflinks和Trinity)进行对isoform进行组装,能鉴定到PacBio的isoform分别仅为22%和8%,表明转录组短读长分析方法在检测isoform的局限性,而PacBio长读长能得到精确的isoform,尤其是在一个基因对应几种isoform的复杂情况下优势明显。
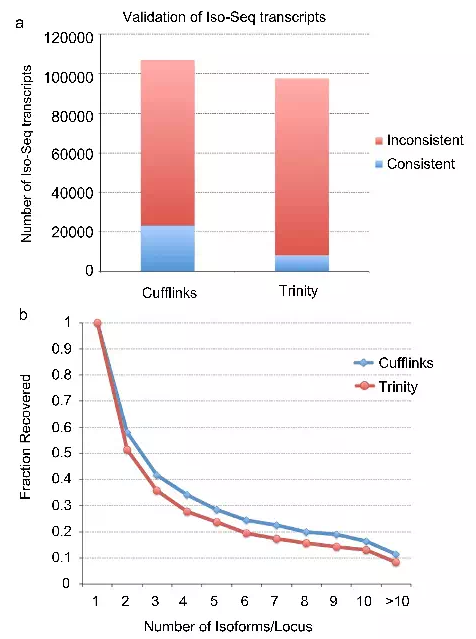 PacBio的isoform对短读长组装构建的isoform评估
PacBio的isoform对短读长组装构建的isoform评估
参考文献
Bo Wang et al., (2016). Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. NATURE COMMUNICATIONS.
官方微信公众号

希望组

希望组科技服务

希望组诊断服务