
产品优势
多样化平台:Nanopore,PacBio,Bionano,Hi-C多种技术平台提供一站式基因组组装服务。
Ultra-long reads:50-100Kb Reads N50,轻松跨越基因组中大片段重复
自主组装算法:NextDenovo,NextPolish自主组装算法可针对不同物种的基因组特点进行优化
云平台急速组装:华为云基因容器超高并发运行测序分析流程,避免任务死锁、减少等待时间,分析速度如虎添翼。
项目经验丰富:国内率先提供三代测序服务,丰富的纯三代动植物基因组de novo 经验,自2016年以来陆续完成了以花生基因组、苹果基因组、籼稻R498基因组近完成图、华夏一号基因组等为代表的基因组项目,发表高水平基因组文章18篇(IF>10)。
方案策略
| 简单基因组 | 复杂基因组 | 超大基因组 | 基因组近完成图 | |
| 定义 | 基因组大小<2Gb 杂合度<0.5% 重复序列比例<50% | 基因组大小>2Gb 杂合度>0.5% 重复序列比例>50% | 基因组大小>10Gb | 基于顶尖策略,组装超完美基因组 |
| 文库类型 | PacBio CLR文库 Nanopore Ligation 1D文库 | PacBio HiFi reads文库 Nanopore Ultra-long reads 文库 | ||
| 测序策略 | 50×NGS+50-80×三代+100×Bionano+100×Hi-C+ | 50×NGS+100×三代+100×Bionano+100×Hi-C | 50×NGS+50-100×Ultra-long reads+100×Bionano+100×Hi-C | Ultra-long reads+HiFi reads+Bionano+Hi-C |
| 承诺指标 | Contig N50>1Mb Scaffold N50>5Mb | 视具体物种而定 | – | – |
分析内容
| 数据产出以及数据质控 | |
| 基因组组装 | 1. 组装 2. 组装评估 |
| 基因组注释 | 1. Repeat注释 2. 基因预测 3. 基因功能注释 4. ncRNA注释 |
| 进化分析 | 1. 基因家族鉴定 2. 特有/共有基因分析 3. 进化树构建 4. 物种分化时间计算 5. 基因家族收缩&扩张分析 6. 直系同源基因鉴定 7. 正向选择基因分析 8. 全基因组复制事件预测(部分物种) 9 大片段复制事件预测(需要构建染色体) |
| 个性化分析 | 根据项目和物种特点设计个性化分析方案 |
结果展示
基因组组装
希望组已经基于纳米孔测序技术完成了多个10Gb以上大型基因组的测序组装工作,凭借高质量的纳米孔长读长数据以及NextDenovo组装算法,获得的大型基因组的Contig N50均在Mb水平,基因组完整性也较高。
| Sample | Heterozygosity (%) | Long reads Data(Gb) | Nanopore Reads N50(Kb) | Assembly size (Gb) | Contig N50(Mb) | Contig Number (#) | BUSCO(%) |
| 某双子叶植物1 | 1.9 | 1,066 | 23.26 | 16.75 | 1.96 | 12,117 | 82.33 |
| 某双子叶植物2 | – | 800 | 26.21 | 10.74 | 12.73 | 5,274 | 83.58 |
| 某单子叶植物1 | – | 1,027 | 50.46 | 10.76 | 93.26 | 329 | 97.75 |
| 某单子叶植物2 | 0.7 | 754 | 30.98 | 10.80 | 5.84 | 3,872 | 80.47 |
| 某裸子植物 | – | 896 | 23.12 | 10.92 | 9.99 | 4,432 | 82.43 |
基因家族分析
采用OrthoMCL对物种进行基因家族聚类分析
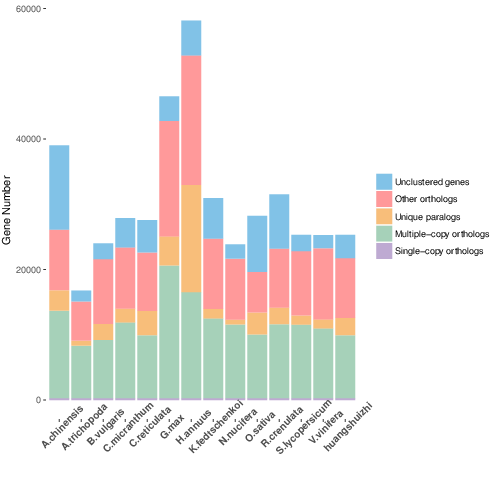
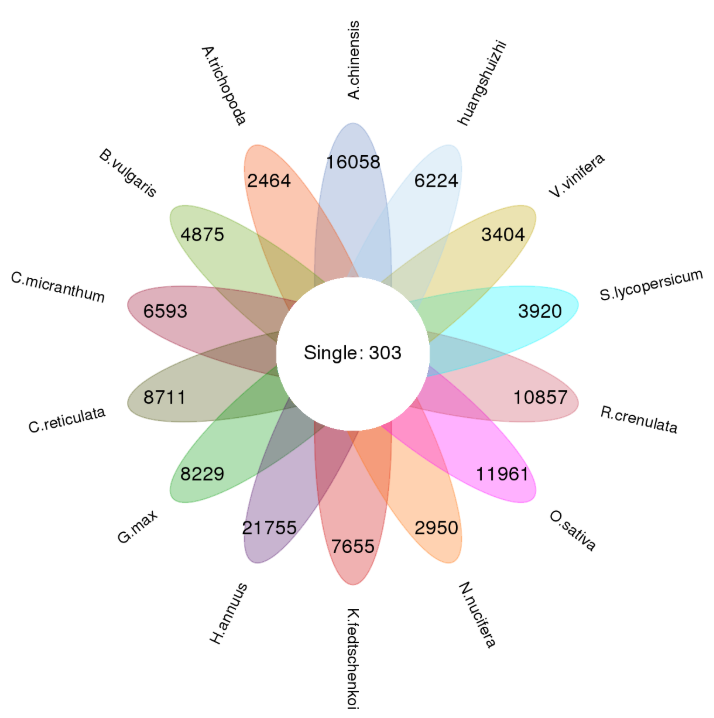 图1 a各样本基因家族类型分类图,b 单拷贝基因与物种本身特有基因花瓣图
图1 a各样本基因家族类型分类图,b 单拷贝基因与物种本身特有基因花瓣图
构建系统进化树
构建系统发育树的过程实际上就是利用同源性状(homologous trait)(例如MSA中的某一列位点)及同源状态(homologous state)推断祖先状态(anceteral state)的过程。
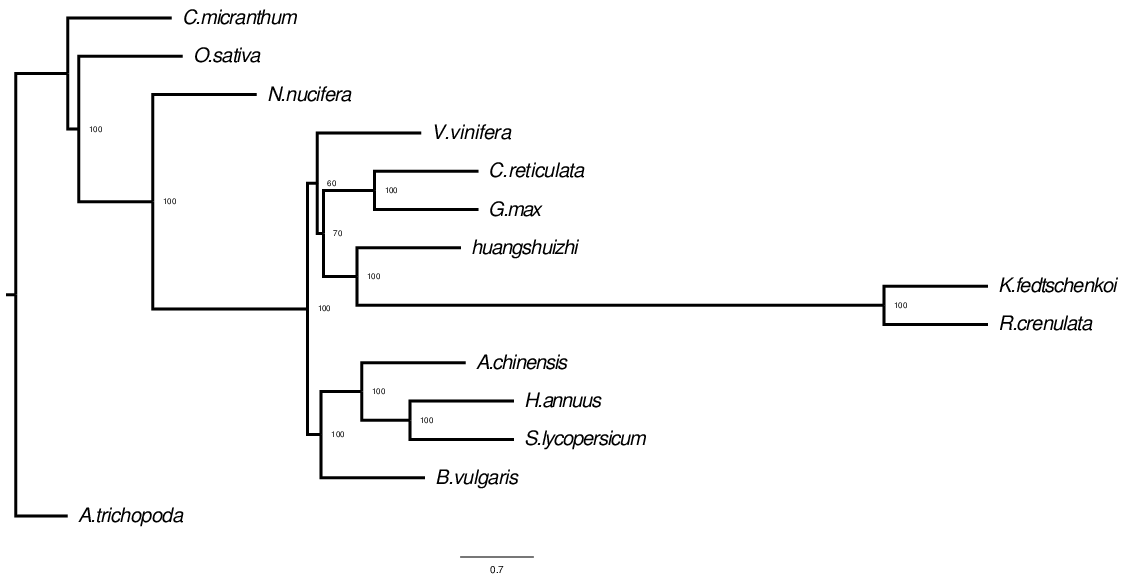 图2 进化树树形结构
图2 进化树树形结构
估算分歧时间
分歧时间(divergence time)是当前进化分析的一个热点,以某一特定类群的化石记录作为参照点,通过基因序列间的分歧程度以及分子钟来估计分支间的分化时间,同时计算系统发育树上其他节点的发生时间,从而推断相关类群的起源时间和不同类群的分化时间。
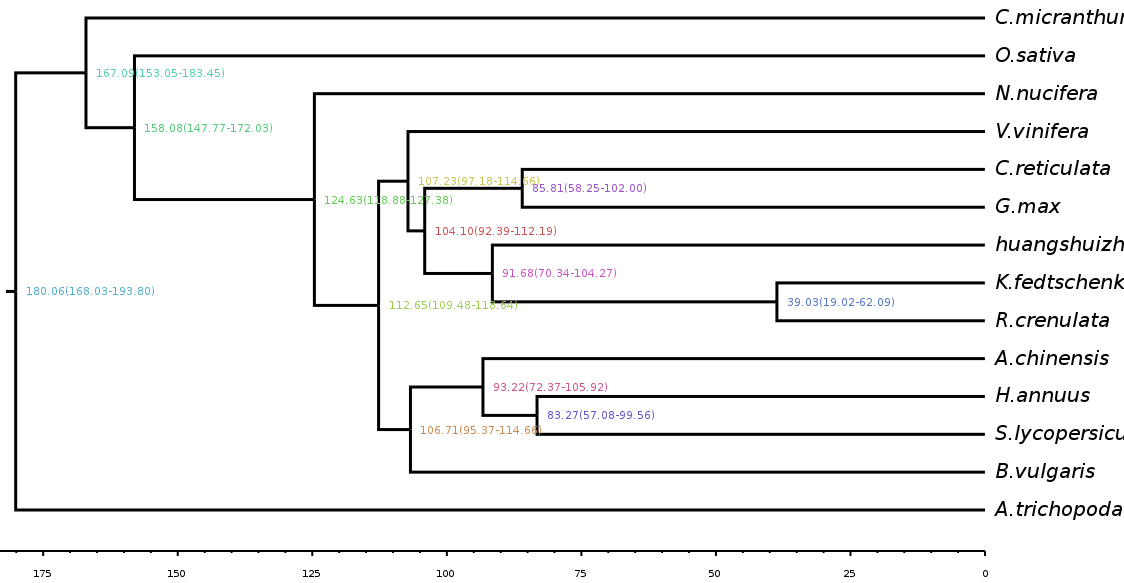 图3 分化时间估算图
图3 分化时间估算图
基因家族收缩扩张分析
物种在进化过程中由于受到各种选择压力,基因会出现扩张和收缩的现象,通过对基因家族的鉴定以及与祖先数据的比较分析,可以得到不同物种的基因家族是否发生了大规模的扩张或收缩甚至丢失,来推断其所受的自然选择压力。利用物种在基因家族中的数目文件和分歧时间,得到不同物种在各个进化分支上的基因家族进化情况。
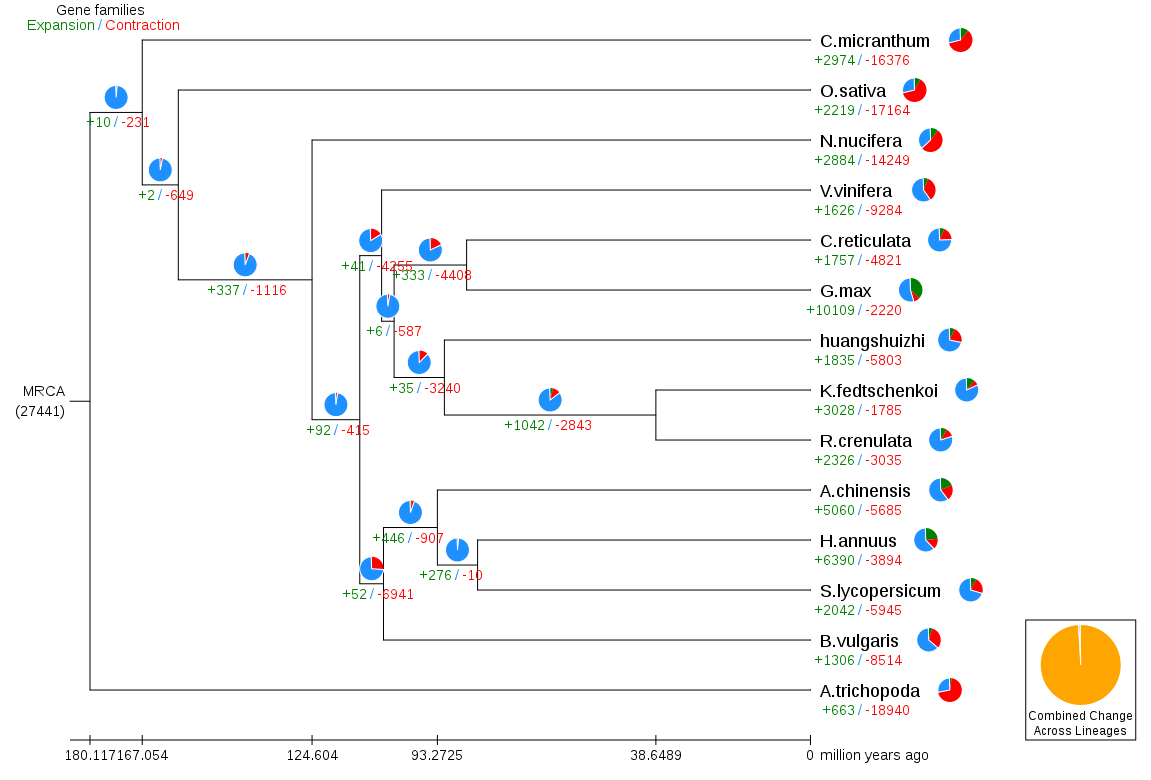 图4 基因家族扩张收缩情况
图4 基因家族扩张收缩情况
全基因组加倍分析
全基因组加倍(Whole Genome Duplication,WGD,又称全基因组复制或全基因组多倍化)是生物演化史上的重要事件,在物种起源、基因组扩张等方面有重要意义。4DTV(four-fold synonymous third-codon transversion)值和同义替换率Ks可以分析物种在进化史中是否发生全基因组复制事件。
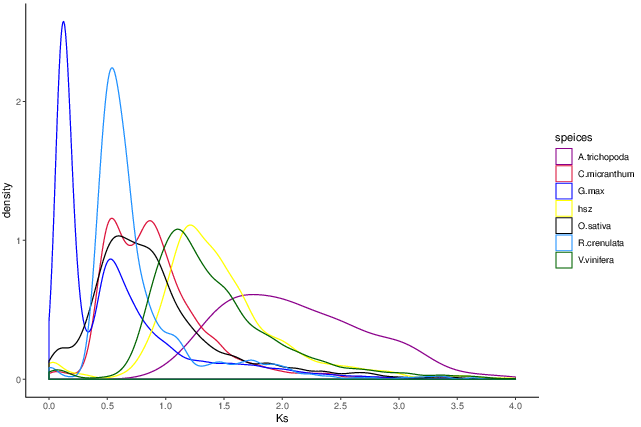 图5 物种Ks分布图
图5 物种Ks分布图
常见问题
1 基因组de novo的组装结果好坏如何判断?
从头组装基因评价通常包含三个方面:连续性、完整性和准确性。
一般用contig N50和scaffold N50 来衡量连续性,N50是指把组装出的contigs或scaffolds从大到小排列,当其累计长度刚刚超过全部组装序列总长度50%时,最后一个contig或scaffold的大小即为N50的大小。BUSCO(Benchmarking Universal Single-Copy Orthologs)在基因含量层面来评估其基因组完整性。序列一致性评估旨在利用高质量的二代测序数据来评估三代测序数据组装结果在单碱基水平上的准确性。
2 PacBio/Nanopore测序优势是什么?
对动植物基因组进行测序及拼接组装(纯三代组装),较好地解决了传统测序组装技术中高杂合度、高度重复区域、异常GC含量区域等诸多组装难题,极大提升了基因组的组装指标,是过去二代测序策略的10-20倍以上。
3 Nanopore Ultra-long reads对基因组组装有什么帮助?
牛津纳米孔测序平台独有的Ultra-long测序能够产生超长测序片段,轻松跨越基因组中连续重复或大片段重复区域,更大限度地还原真实的基因组景观。
轻松跨越重复区域
对于基因组中“暗区”,二代测序短读长无法跨越,三代测序普通文库不能完全解决,而Ultra-Long Reads能够轻松跨越连续重复区域,提供更多的序列信息,更便于组装过程重复片段划分。
显著提升组装质量
在基因组组装过程中可以通过增加读长获得理想组装质量,加入Ultra-Long Reads数据可以显著提升人类基因组组装效果,填补基因组中的缺口,甚至组装出端粒到端粒水平的完整染色体。
案例解析
案例分析一
案例解析
四倍体栽培种花生基因组揭示豆科核型,多倍体进化及作物驯化的秘密
栽培种花生(Arachis hypogaea L.)为异源四倍体(AABB,2n = 4x = 40),其亚基因组之间的密切关系和高比例的重复序列增加了栽培花生基因组的组装难度。本研究在全世界范围内首次破译了四倍体栽培种花生的全基因组,为花生染色体起源、花生及豆科主要类群核型演化、花生基因组结构变异、花生物种起源与分子育种等研究提供新思路。
测序、组装及注释
本研究以狮头企(Arachis hypogaea var. Shitouqi)花生为材料,对100x PacBio数据进行初步组装(平均读长10.25Kb),获得Contig N50为1.51Mb。接着利用Hi-C数据将组装结果提升至染色体水平,N50为129.8 Mb。最后利用高密度遗传图谱对5个含有轻微组装错误的Hi-C结果进行调整,最终组装出大小2.51Gb,包含20条染色体的四倍体栽培种花生基因组。为了评价组装效果,与公布的花生BAC双末端测序数据、三个花生全长BAC序列比对都显示高度的一致性,另外通过二代测序数据和三代的测序数据进行了碱基水平的准确性评估和连续性评估,所有的评估结果表明了花生基因组高质量组装。从花生基因组中共鉴定到30,596个非冗余基因,24,208个同源基因对在两个亚基因组之间表现出广泛的差异表达,其中B亚组的显性表达频率高于A亚组。
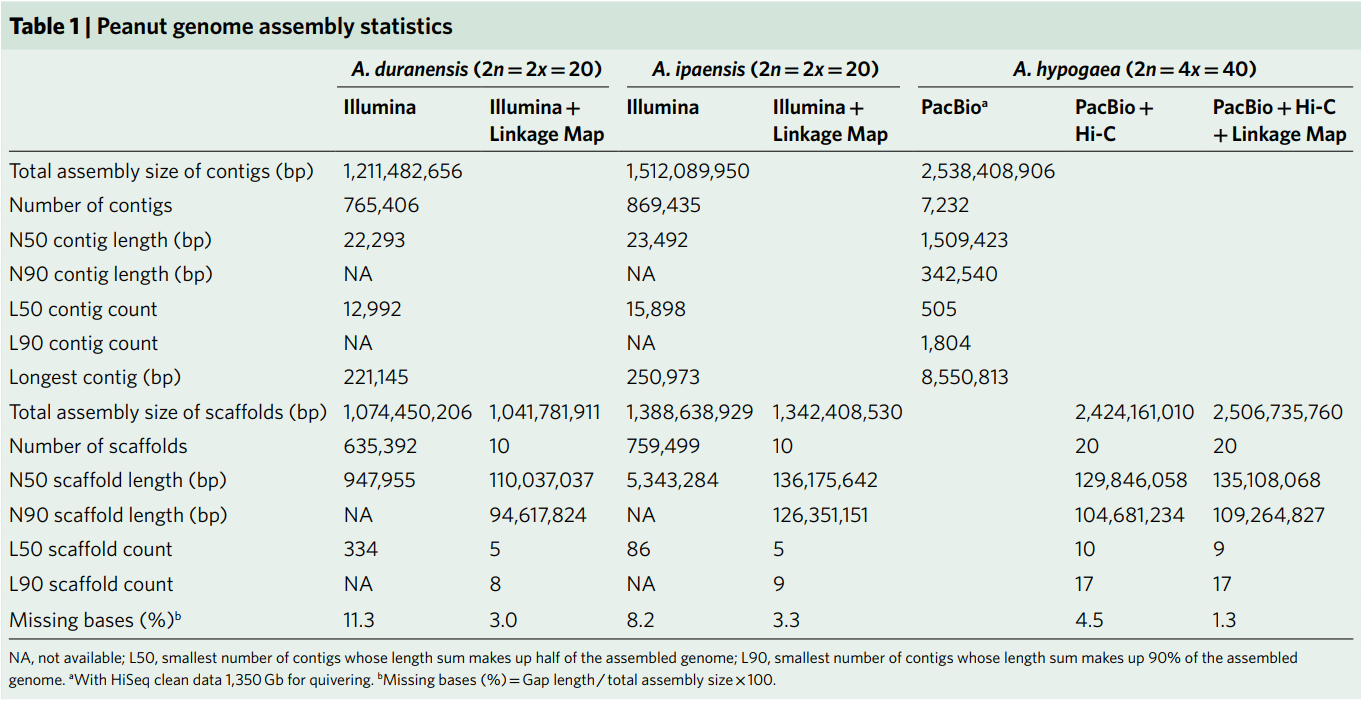
表1 花生基因组组装统计
亚基因组结构特征
比较基因组结果表明花生栽培种B亚组与二倍体A. ipaensis一致性高于A亚组与A. duranensis之间的一致性。共有629个基因受到基因转换的影响,有58.7% B转换为A,41.3% A转换为B。A和B亚组之间存在较多的倒转和重组,鉴定到至少6个有明确界限的A、B亚基因组之间的交换或替换,包括染色体3和13之间的10Mb易位。
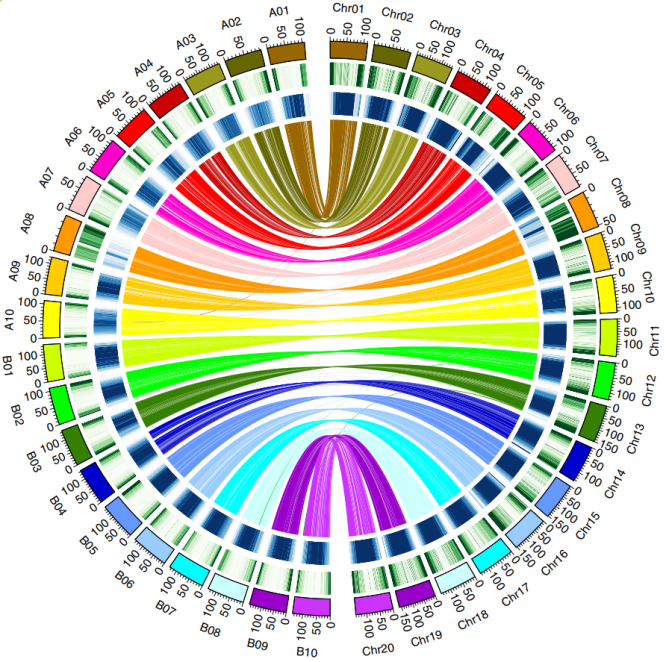 图1 花生亚基因组与二倍体A、B基因组基因密度、重复序列共线性关系
图1 花生亚基因组与二倍体A、B基因组基因密度、重复序列共线性关系
通过完整的LTR反转座子两端的LTR序列进行比对计算碱基替代率表明A亚基因组在四倍体化后(约25万年前)经历了快速的LTR扩增,而B亚基因组和两个二倍体的LTR在四倍体化前扩增,这可能是由于功能障碍表达的普遍存在或四倍体花生中亚基因组同源染色体的缺失造成的,作者在这里提出疑问:测序的二倍体野生花生A. duranensis是否就是A亚基因组的祖先?
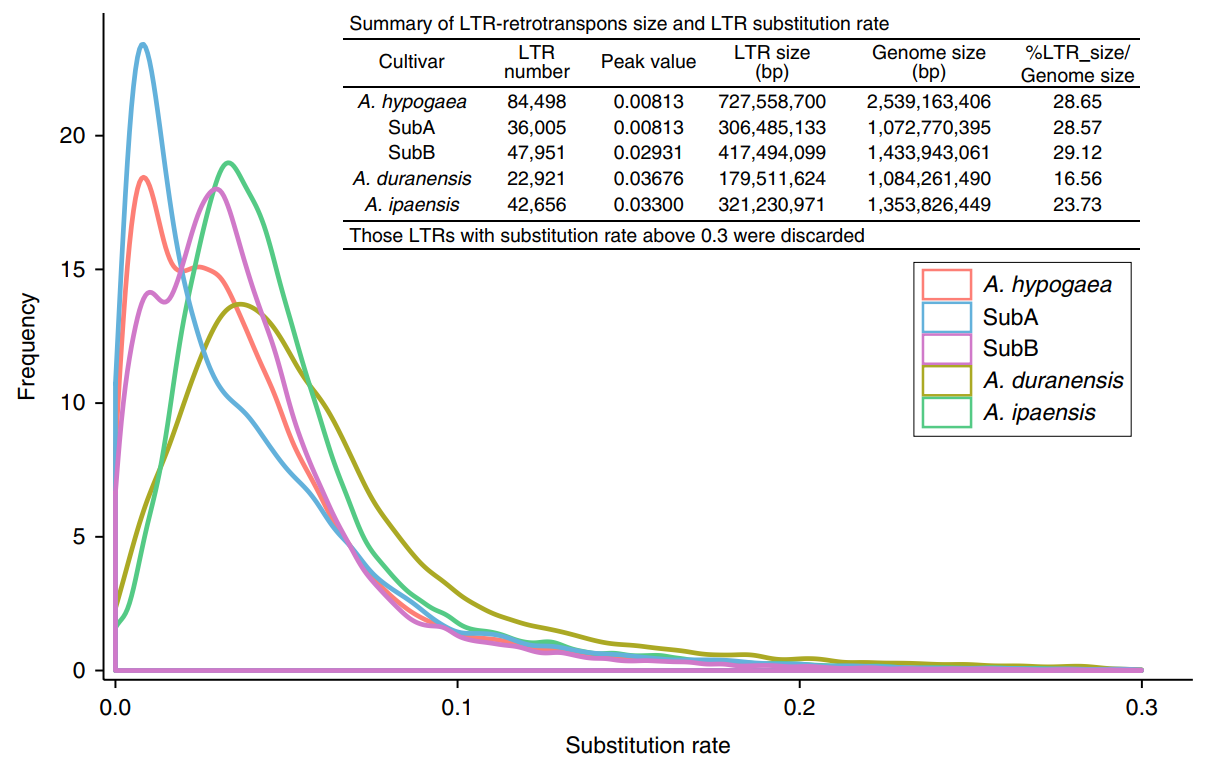 图2 花生及其二倍体祖先的重复序列扩增
图2 花生及其二倍体祖先的重复序列扩增
豆类植物共有的四倍化(legume-common tetraploidy,LCT;约5900万年前),以及主要双子叶植物共有的六倍化(core-eudicot-common hexaploidy,ECH;约1.3亿年前)痕迹保留在花生基因组中。作者利用保留有Post-ECH和post-LCT的普通豆类基因重建了16条原始豆类染色体(称为Lu),与现存的豆类基因组进行比较并绘制了花生与其他豆类的核型进化图,推断花生染色体的形成过程。花生祖先染色体A1,A3,A4,A5,A6和A7由Lu染色体经过6次融合造成染色体数目减少的片段组成;而A2,A8,A9和A10由两条Lu染色体的交叉互换产生;从A基因组分离以后,B基因组内的交叉互换形成了其特有的7、8号染色体。
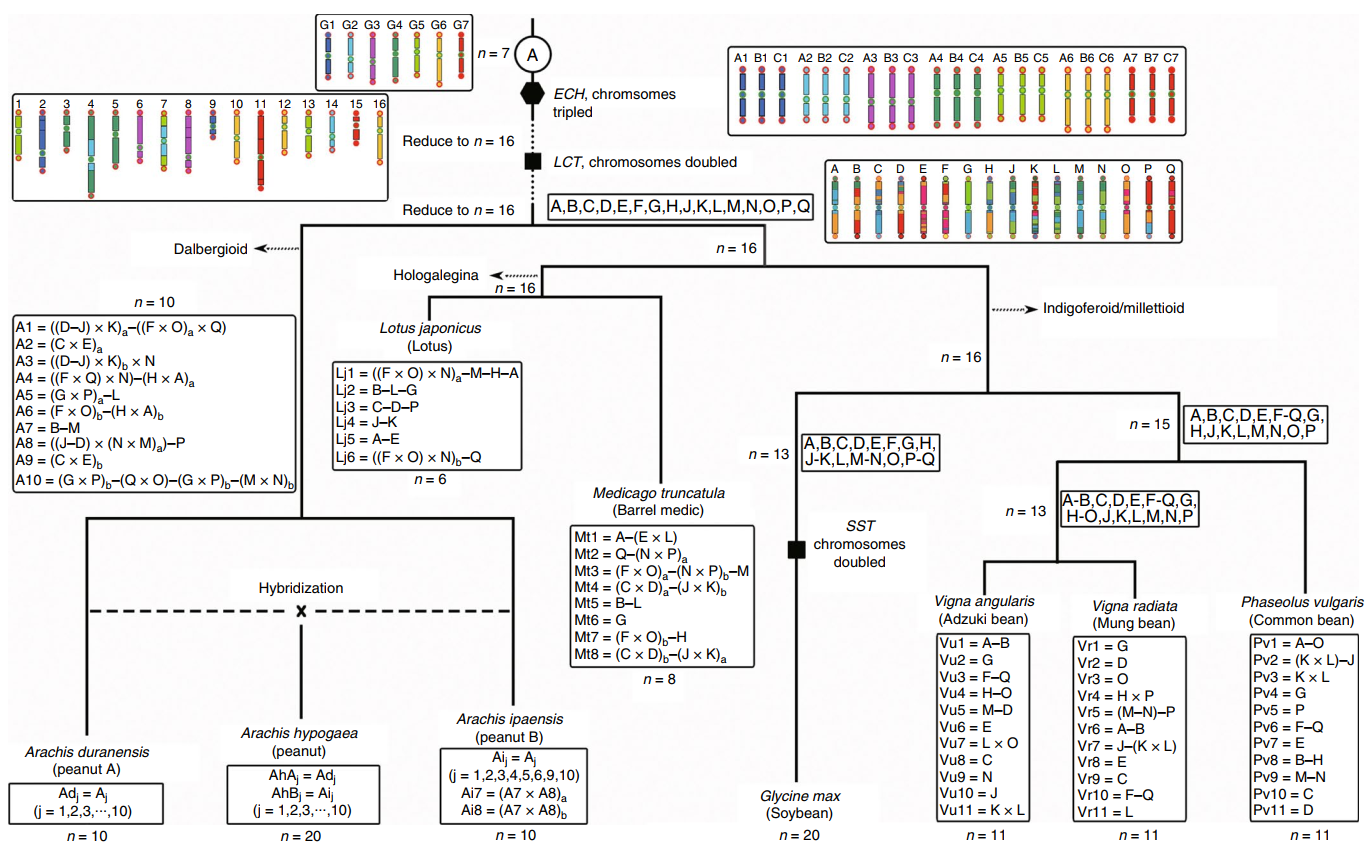 图3 花生与其他豆类的核型进化图
图3 花生与其他豆类的核型进化图
亚基因组含量变化
与二倍体花生A. duranensis和A. ipaensis相比,四倍体花生亚基因组A(37,059 genes)和B(46,650 genes)分别有0.88%和12.46%的扩张,在A和B基因组二倍体中鉴定的24,380个同源基因家族中,90.68%在四倍化后仍旧保留。四倍体花生、野生A基因组和野生B基因组中的生长素响应因子(ARF)分别有114、28和28个,聚类为9个簇,其中Ⅰ-V仅包括四倍体花生的拷贝,同时花生含有3个CYP78A6(与种子生长有关),而二倍体B基因组中仅有一个拷贝,这可能与花生籽粒大小有关。驯化过程中同样会出现基因丢失的现象,例如四倍体花生有661个NBS结构的抗病基因,总数少于A. duranensis(385)和A.ipaensis(428)的总和,造成四倍体花生抗病基因的减少。作者还构建了花生基因组水平的酰基-脂质代谢网络和共生(SYM)信号通路基因的系统发育树,为花生品质改良及固氮研究提供支持。
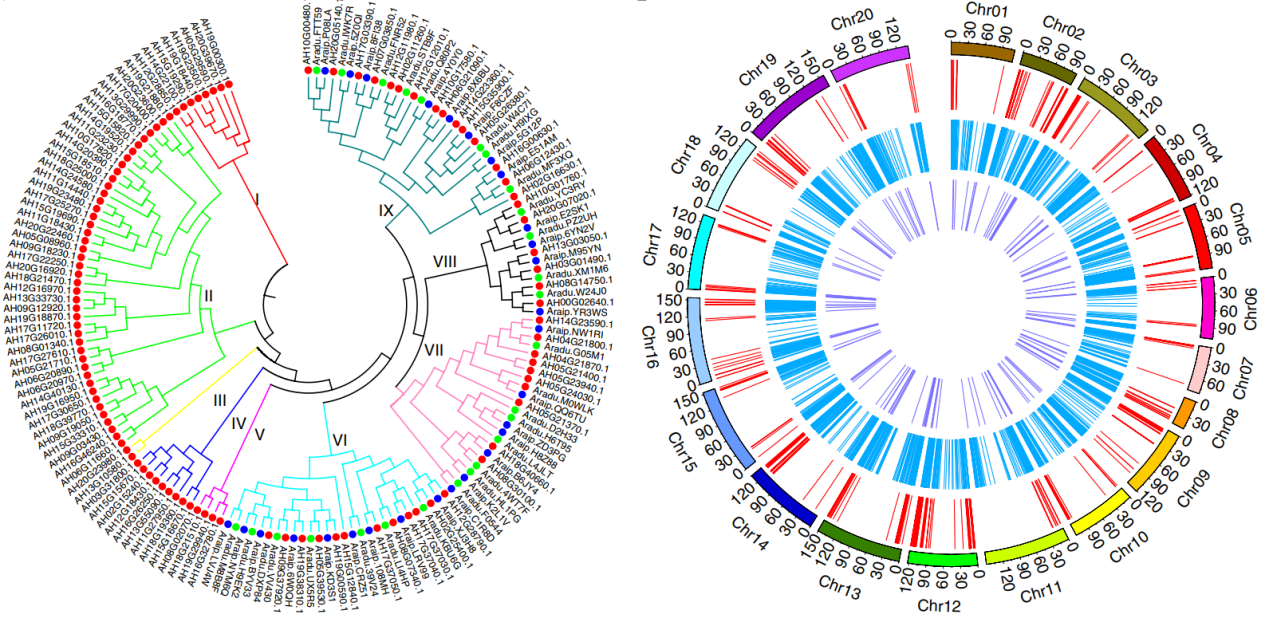 图4 生长素响应转录因子(ARF)家族进化树及脂肪酸代谢、氮共生途径及抗病基因染色体分布
图4 生长素响应转录因子(ARF)家族进化树及脂肪酸代谢、氮共生途径及抗病基因染色体分布
花生的起源和驯化
为了研究花生的起源和驯化,作者构建了52份样品(30个不同生态型异源四倍体花生,18个野生种,4个合成四倍体)的系统发育树(图5b)。系统发育树及测序数据表明野生型四倍体A.monticola形成了subsp. hypogaea和fastigiata生态型,这表明花生可能起源于不同的subsp. hypogaea并且在不同地点独立驯化,例如秘鲁西北地区进化出适应干旱的生态型(图5d,箭头B),东南独立驯化产生的瓦伦西亚和西班牙生态型在世界范围传播(图5d,箭头C和D)。这有别于前人预测的花生由A. monticola在阿根廷北部驯化而来。
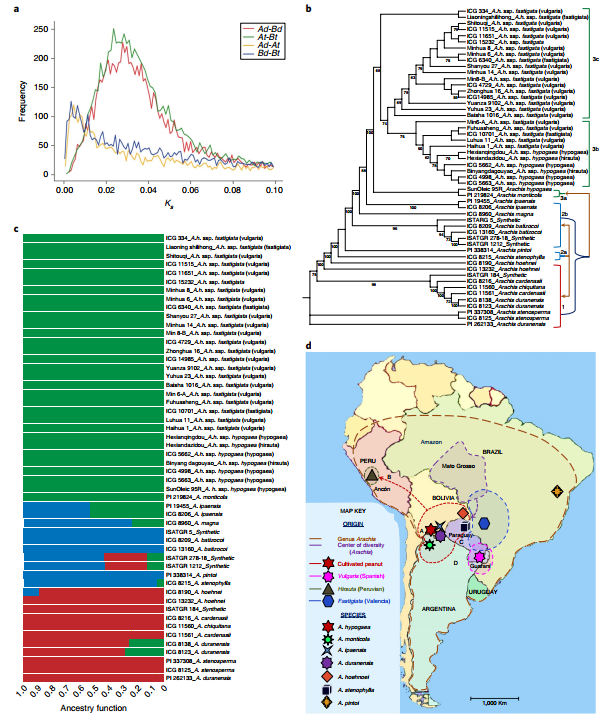 图5 花生的进化历史
图5 花生的进化历史
四个合成四倍体中ISATGR 278和ISATGR 5发生了全基因组的加倍,而另外两个的A基因组分别是B基因组的1.23和5.93倍,这可能是由于亲本染色体由于不相容而在后代中不随机保留,这进一步支持了作者的假设:存在另一个与B基因组更相容的A基因组供体,而不是A. duranensis。
对花生性状改良的影响
该基因组揭示了许多已被基因定位的花生重要农艺性状的候选基因。控制红色种皮的单显性基因定位到3号染色体上一段0.905cM的区间内,包含WRKYs, MYB和bHLH家族以及细胞色素P450等花青素合成相关基因,这些基因的上调表达可能是红色种皮形成的原因。花生种子大小是重要的产量指标,作者利用一个重组自交系群体结合BSA分析在chr07和chr12染色体上定位到两个相同的候选区段,分别包含99和97个候选基因。
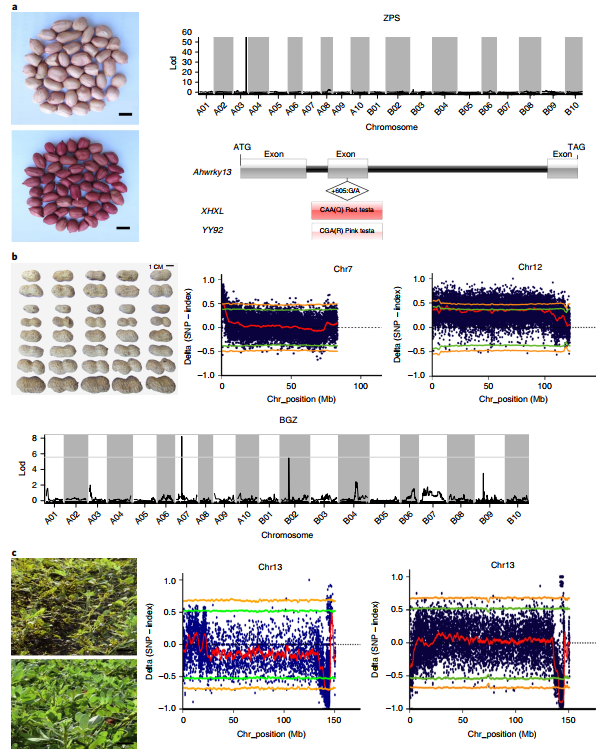 图6 种子大小、颜色和叶片抗病性的候选基因
图6 种子大小、颜色和叶片抗病性的候选基因
花生叶锈病和晚叶斑病(late leaf spot, LLS)共定位在同一基因组区域,重组自交群体抗病和感病池在Chr13染色体上显示重叠区域,进一步分析表明该区段内保守的Tir-NBS-LRR基因AH13G54010.1可能是两种病害的抗病基因。从含油量约40%的材料中获得了含油量高达80%的突变系,并通过重测序结合四倍体组装基因组解释了高含油量是由于ahFAD2A和ahFAD2B两种突变共同引起的。
参考文献:
Zhuang WJ, Chen H, Yang M, et al., The genome of cultivated peanut provides insight into legume karyotypes, polyploid evolution and crop domestication. Nature Genetics. 2019.
https://doi.org/10.1038/s41588-019-0402-2
官方微信公众号

希望组

希望组科技服务

希望组诊断服务