
希望组与天津生物芯片两家公司强强联手,结合各自多年在动植物基因组组装方面的实战经验、整合双方优势资源,联合首推Nanopore Ultra-long Reads+PacBio HiFi Reads+Bionano+HiC的基因组近完成图解决方案,突破现有基因组技术指标,为客户提供更有效、更完整、更准确的基因组图谱,为后续基因组进化、基因组结构变异、基因功能研究等更深入的研究奠定基础。
Nanopore Ultra-long Reads组装,透视基因组复杂区域
现有常用策略组装的动植物基因组通常都是不完整的,最主要的原因是基因组中重复序列太长(尤其是着丝粒和端粒区域),而测序Reads太短难以跨越重复区域。三代测序相比二代测序在读长方面有了极大的提升,但是仍有较多长重复区域的组装错误[1]。Nanopore Ultra-long Reads是现有技术中解决这一问题的最佳方法之一,Ultra-long Reads N50可达100Kb以上能够有效跨越基因组中上百Kb的大片段重复,甚至着丝粒区域[1,2,3]。
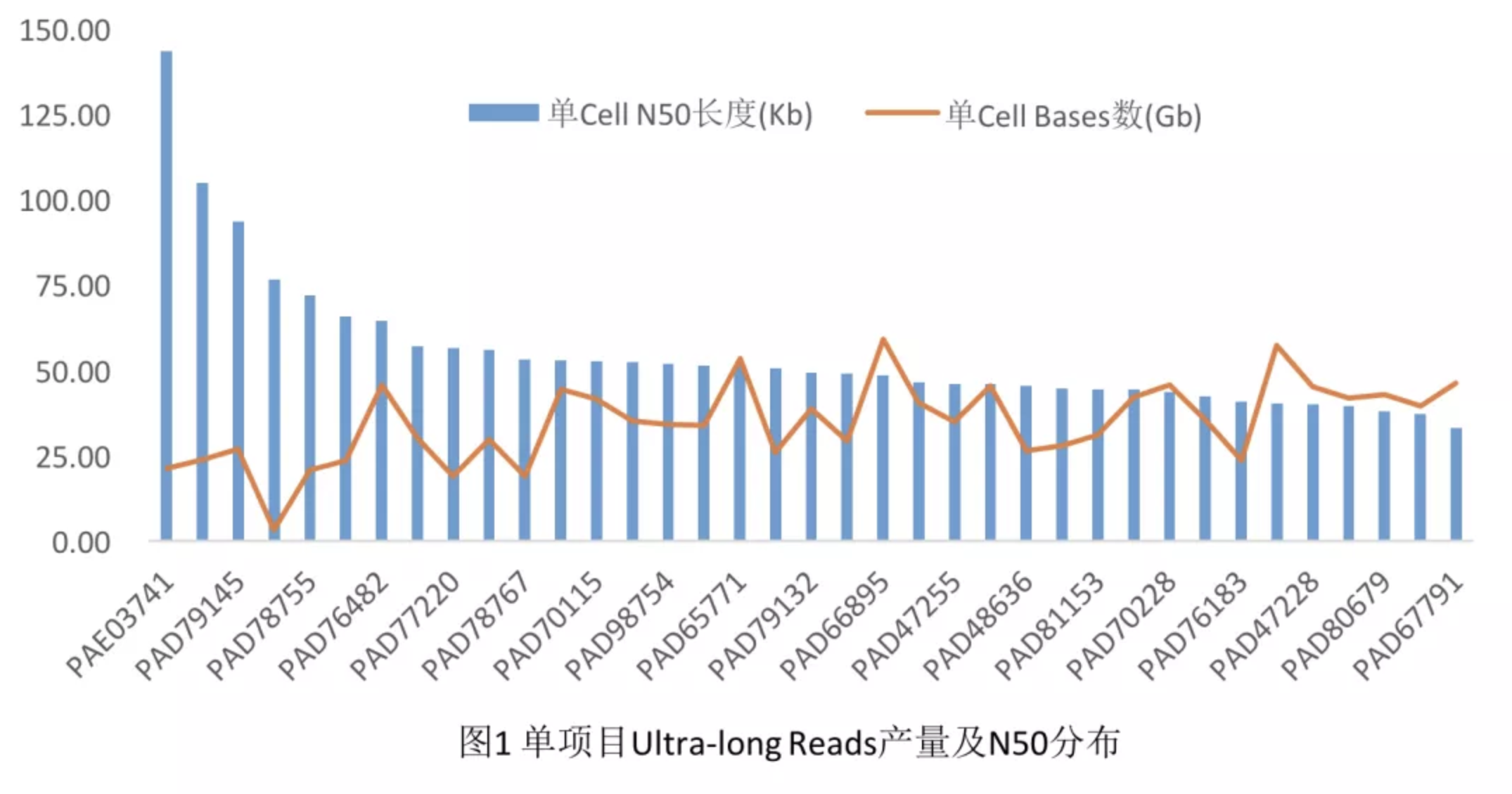
据悉,希望组是目前国内少数能稳定产出高质量Nanopore Ultra-long Reads的服务商之一(图1),基于Ultra-long Reads的动植物基因组组装经验丰富。例如,某基因组大小约10G的单子叶植物,采用Ultra-long Reads组装contig N50达93Mb(表1)。Nanopore Ultra-long Reads组装,透视基因组复杂区域,为基因组近完成图打下坚实的基础。
表1 某单子叶植物基因组Ultra-long Reads组装结果
| StatType | Contig Length(bp) | ContigNumber |
| N50 | 93,262,735 | 34 |
| N60 | 75,353,051 | 47 |
| N70 | 60,156,522 | 63 |
| N80 | 43,730,326 | 84 |
| N90 | 20,933,943 | 119 |
| Longest | 405,550,188 | 1 |
| Total | 10,759,349,041 | 329 |
| Length>=1kb | 10,759,349,041 | 329 |
| Length>=2kb | 10,759,349,041 | 329 |
| Length>=5kb | 10,759,349,041 | 329 |
PacBio HiFi Reads纠错提升单碱基准确性
Nanopore Ultra-long Reads解决了基因组复杂区域的组装问题,但只有近完成图的底子,其单碱基准确度仍略显不足。目前主流的解决方法是利用二代测序数据进行Polish,但是二代测序在基因组高GC区域有偏好性,产生的短读长不能均匀覆盖基因组,Polish后的基因组仍有许多单碱基错误和小InDels[1]。在实际项目经验中,研究人员发现采用PacBio HiFi Reads对基因组Polish效果更好。
PacBio 升级至Sequel II后HiFi Reads单碱基准确性有了极大提升,并且HiFi Reads测序过程没有GC偏好性,覆盖基因组更加均匀,避免了短读长的多位点比对错误,Polish后的基因组准确度更高。
天津生物芯片自2013年开展PacBio测序至今,数年来一直深耕PacBio样品处理、文库构建、上机测序及数据组装技术,目前Sequel II 平台采用最新的2.0试剂,HiFi模式 Total Bases超过了400Gb,CCS Bases 达到了30Gb以上。PacBio HiFi Reads不仅能够De novo组装,还能够解决高杂合度基因组Phasing问题,以及实现高质量基因组的Polish。同时,高通量的测序数据产出,能够有效降低测序成本,节省纠错时间,缩短组装周期,成为获得动植物近完成图基因组不可或缺的重要手段。
表2 测序及组装结果统计(某高杂合木本植物基因组)
| PacBio Sequel II | Polymerase N50 | Subread N50 | Depth | Assembled genome size | contigs N50 | BUSCO assessment |
| 405 Gb | 186,887bp | 20,409bp | 468× | 865.3Mb | 23.53Mb | 98.35% |
NextDenovo+NextPolish自主产权组装算法突破技术瓶颈
工欲善其事,必先利其器。不同物种的基因组组成各有特点,基因组近完成图组装过程也会遇到各种问题,如果没有强大的组装软件,Ultra-long Reads与HiFi Reads仅仅是一堆数据。NextDenovo是希望组自主研发的三代测序数据高效纠错、组装软件,解决了现有三代测序数据组装工具资源占用大、运行时间长、组装质量不稳定的瓶颈。NextDenovo已经实现了单Contig组装一条染色体的突破,组装的水稻93-11(Oryza sativa L. 2n=24)基因组仅包含18条 Contigs,至少有一半的单条染色体由单个Contig装出(图2)。NextPolish[4]是希望组开发的解决Nanopore测序数据组装基因组准确度的工具包,运行性能优于现有主流三代基因组Polish工具。希望组自主组装算法可以针对不同物种的基因组特点进行优化,是实现基因组近完成图的核心工具。
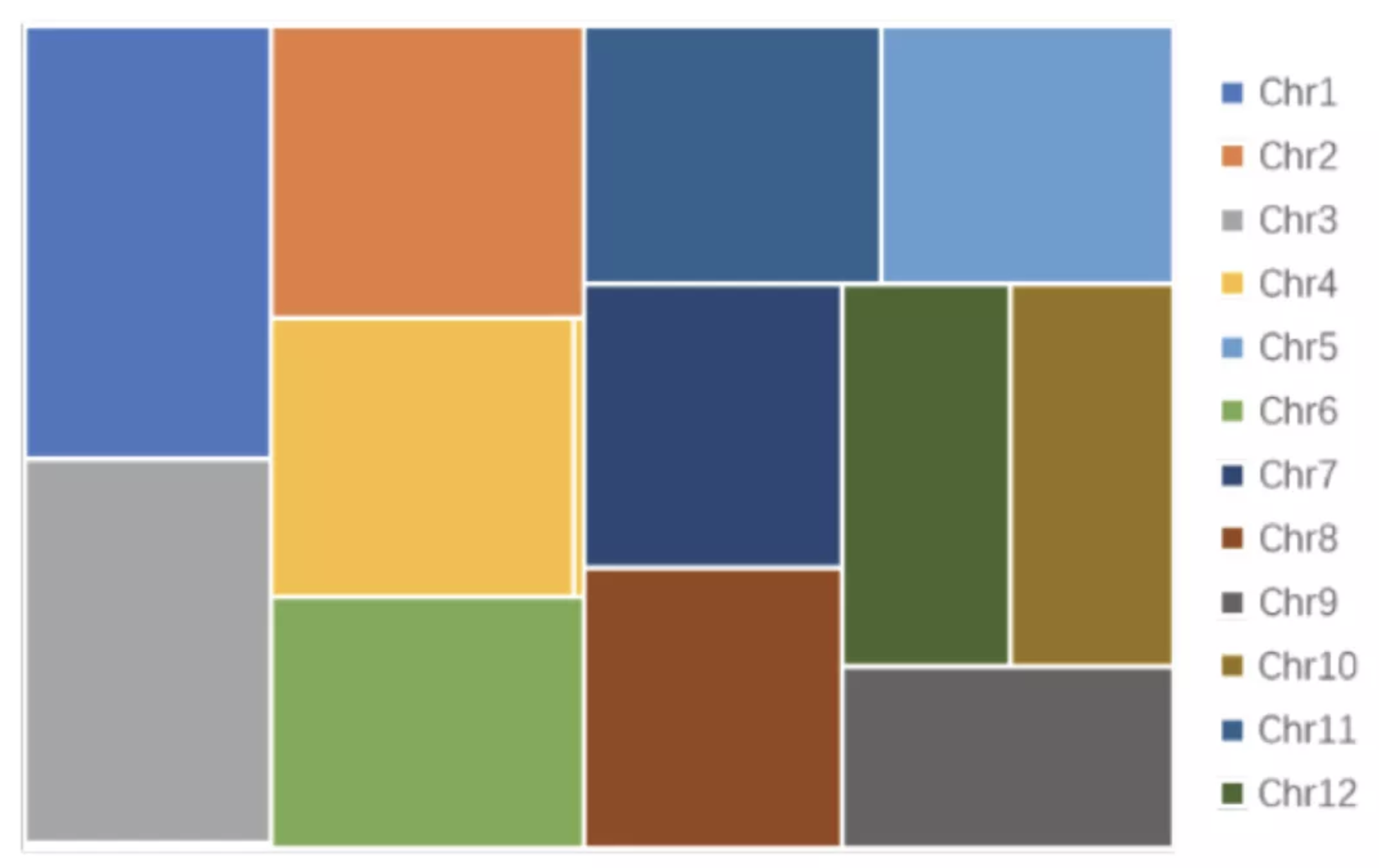

图2 利用NextDenovo组装水稻93-11染色体树状图
Bionano物理图谱校正,组装质量更上一层楼
Bionano 光学图谱技术是基于DNA中一些可识别的限制性酶切位点在DNA上的物理位置而构建的图谱。该技术无片段化操作,无PCR过程,反映DNA最真实的信息,改善了基因组结构的可视化,以最连续、最精确的组装获得染色体臂和完整的染色体图谱。在Ultra-long Reads组装+HiFi Reads Polish的基础上,利用Bionano数据矫正和验证基因组序列组装的的准确性和完整性,从而使组装质量更上一层楼(表3)。
表3 希望组Bionano辅助组装案例
| 物种 | 有效深度 | 辅助组装前(Contig N50) | 辅助组装后(Scaffold N50) | 组装提升效果 |
| 某动物 | 74× | 10.2Mb | 63.8Mb | 6.25倍 |
| 某植物 | 40× | 1.5Mb | 43.6Mb | 29.1倍 |
| 某昆虫 | 41× | 2.8Mb | 25.4Mb | 9倍 |
| 某植物 | 100× | 2.7Mb | 24.4Mb | 9倍 |
Hi-C染色质构象捕获助阵为近完成图“画龙点睛”
没有达到染色体水平,怎敢称为基因组近完成图?Hi-C技术是是基因组近完成图最后的点睛之笔。Hi-C染色质构象捕获根据染色质片段间的交互强度随距离衰减的规律,利用高通量测序技术,获得整个染色质三维空间互作关系[5],将scaffolds基因组序列进行染色体群组的划分、排序、定向,对组装准确性进行再次校正,将组装提升到染色体水平。
宝剑锋从磨砺出,只有经过Ultra-long Reads+HiFi Reads+Bionano+HiC的磨砺,基因组近完成图才名副其实!邮件联系sales-support@grandomics.com或希望组当地销售顾问,快人一步尝鲜新策略!
关于希望组
北京希望组生物科技(集团)有限公司(Grandomics Biosciences Co.,Ltd.)成立于2014年,为农业科研、医学科研、临床诊断等领域提供基于三代测序技术的全方位服务。近年来,公司聚焦“科技服务”、“诊断服务”、“生物信息”、“诊断产品”四大业务模块,在武汉建设4,500平米三代测序中心和医学检验实验室,引进国际先进的Oxford Nanopore PromethION 48&beta、PacBio Sequel、MGISEQ、Bionano光学图谱、Hi-C染色体构象捕获等技术平台,在华为云部署高性能计算集群,为客户提供优质的第三代测序、数据分析和存储服务。希望组秉承“科学的边界、技术的极限、伦理的底线、人文的关怀”的理念,致力于打造中国三代测序技术应用领域的第一品牌。
关于天津芯片
天津生物芯片技术有限责任公司(TBC)成立于2003年9月,由天津中新药业集团股份有限公司,南开大学,天津市创业投资有限公司,天津经济技术开发区国有资产经营公司共同出资组建,主要从事微生物检测芯片的研发、基因组学和功能基因组学研究,是国内最早开展大型生物基因组测序的单位之一。2013年,天津生物芯片率先在国内提供三代测序服务,目前已积累丰富的项目经验,成为中国三代测序技术应用的领跑者。
参考文献:
- Lang D, Zhang S, et al.Comparison of the two up-to-date sequencing technologies for genome assembly: HiFi Readsof Pacbio Sequel II system and ultralong reads of Oxford Nanopore,bioRxiv 2020.02.13.948489; doi: https://doi.org/10.1101/2020.02.13.948489.
- Miga K H, Koren S, Rhie A, et al. Telomere-to-telomere assembly of a complete human X chromosome[J]. BioRxiv, 2019: 735928.
- Jain M , Koren S , Miga K H , et al. Nanopore sequencing and assembly of a human genome with ultra-long reads[J]. Nature Biotechnology, 2018.
- Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics btz891 (2019) doi:10.1093/bioinformatics/btz891.
- Burton, J., Adey, A., Patwardhan, R. et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat Biotechnol 31, 1119–1125 (2013). https://doi.org/10.1038/nbt.2727
官方微信公众号

希望组

希望组科技服务

希望组诊断服务