
产品优势
超长读长,轻松获取全长转录本序列,显著降低多位点比对率
大多数传统测序方法需要进行片段化,准确组装完整的转录本仍然存在困难,尤其是在一个读长可以比对到多个位点的情况下(例如高度保守的序列段)。由于 ONT 测序的长读长优势,能够轻松测序得到从 5′ 端到 poly(A)的全长转录本序列,显著减少reads的多位点比对情况,让reads与参考基因组的比对更加准确。
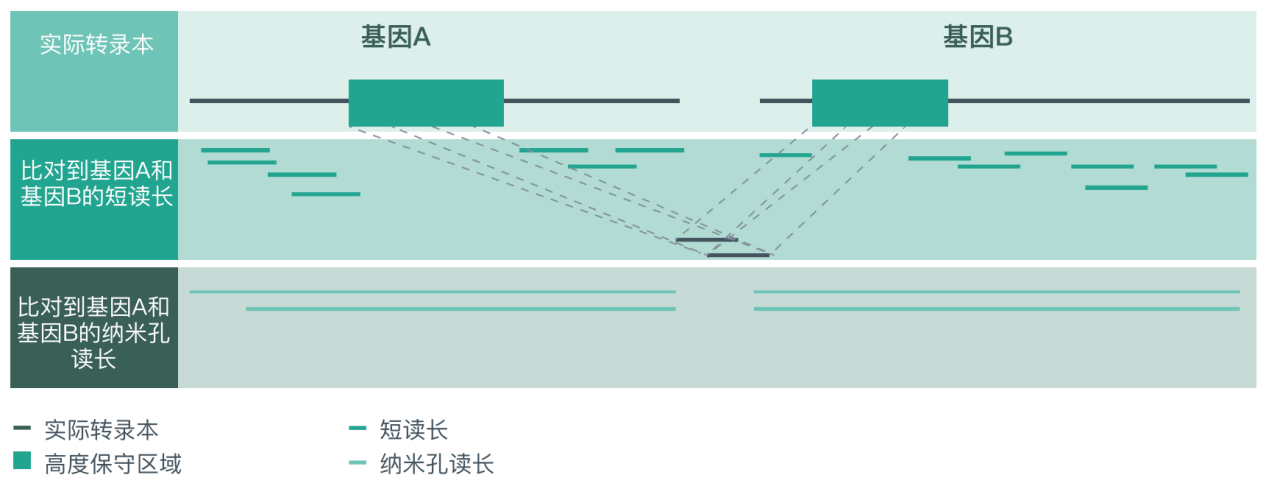
利用短读长和 Nanopore 长读长组装全长转录本
完全覆盖剪接位点,精准识别可变剪接事件
真核生物基因转录时,一个 mRNA 前体通过不同的剪接方式产生不同的 mRNA 剪接异构体的过程称为可变剪接(或选择性剪接,alternative splicing, AS)。选择性剪接可以让每个基因产生大量的 mRNA 异构体,从而促进了基因组成和功能的多样化。传统 RNA 测序技术产生的短读长丢失了位置信息,使得正确组装选择性剪切 mRNA 异构体存在挑战。纳米孔长读长能够横跨剪切位点,从而精准识别可变剪切事件直接对转录本进行表达量化分析。
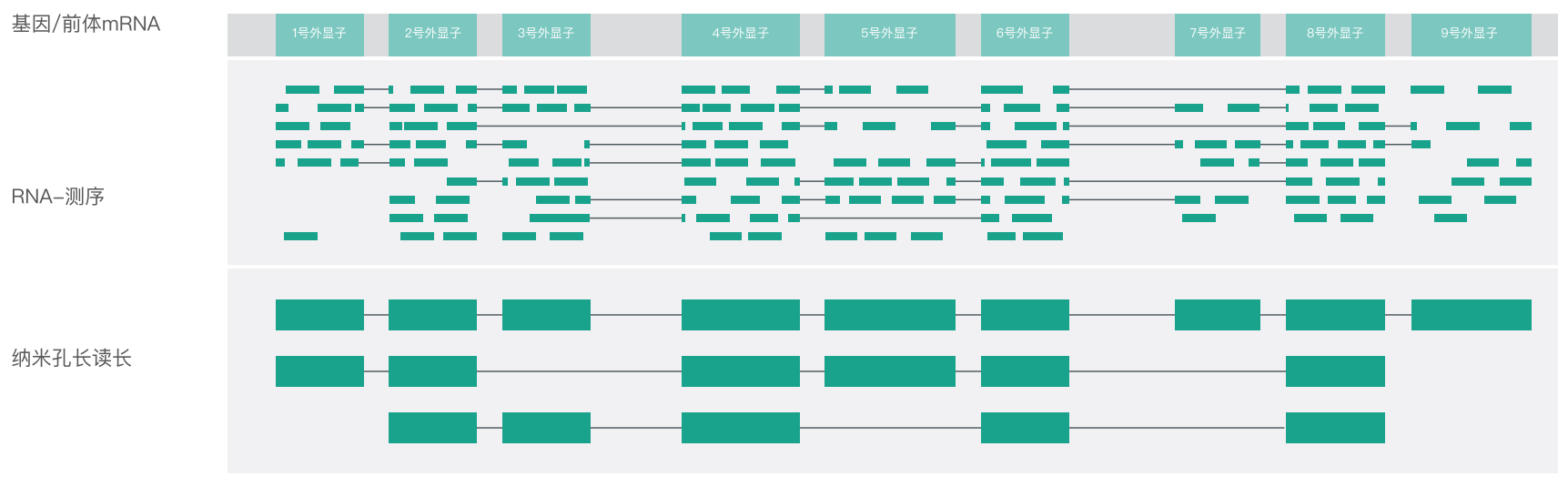
利用短读长和 Nanopore 长读长组装全长转录本
对转录异构体进行表达量化分析
短读长测序由于读长较短,多位点比对率高,无法准确鉴定转录本的异构体,故而不能在转录本水平获得准确的表达量结果。而ONT 转录组测序读长够长,直接获取转录异构体序列,同时由于其足够高的通量,测序可以达到饱和,因此在基因水平和转录本水平都能进行准确的表达量分析。ERCC spike-in 分析显示所有类型 ONT 转录组测序文库都是可定量的。
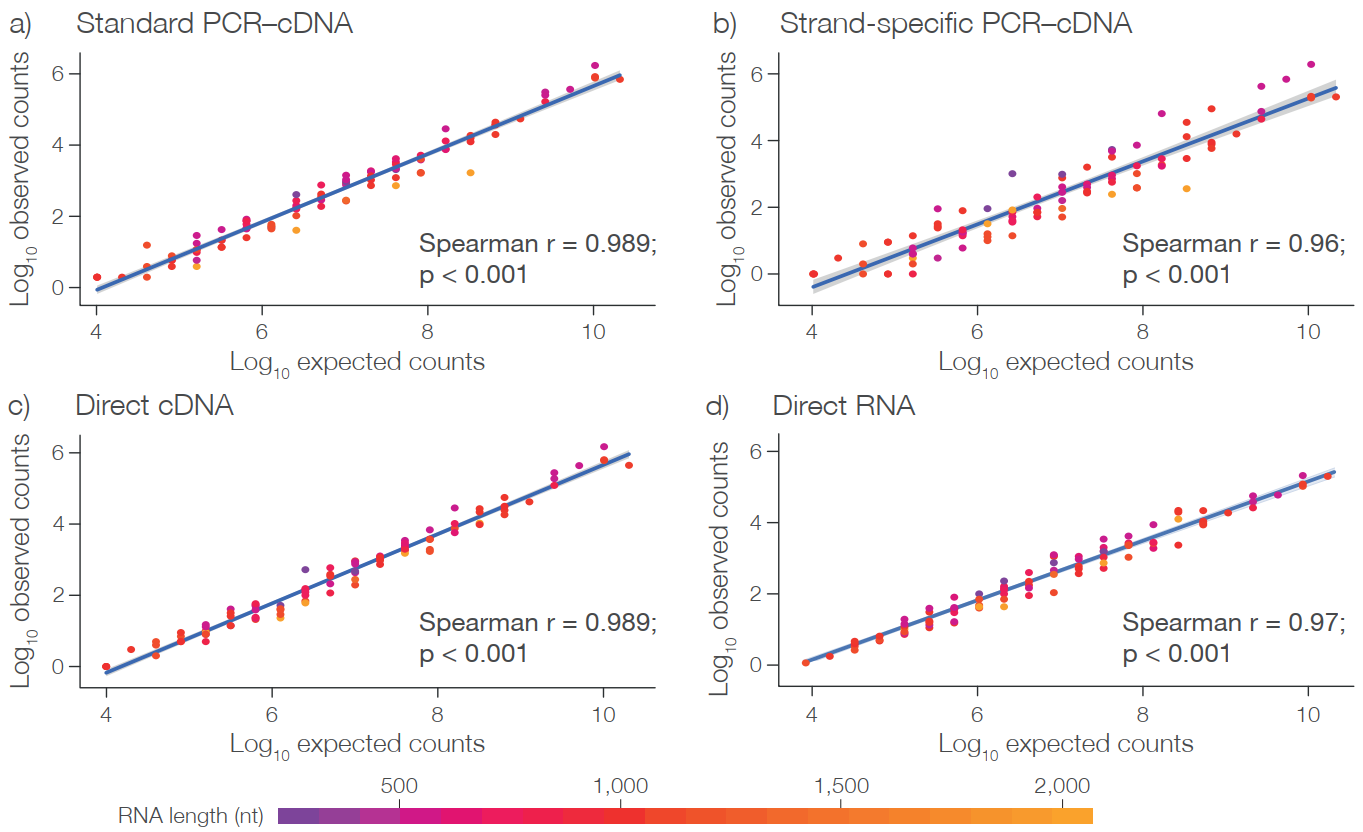 Nanopore 全长转录组定量分析
Nanopore 全长转录组定量分析
方案策略
| RNA样本量 | 建库策略 | 测序策略 |
| 总量 ≥ 0.5 μg, 浓度 ≥50 ng/μL | cDNA PCR扩增后1D建库 | 需基因定量:ONT测序数据量 ≥ 2 Gb |
| 总量 ≥ 0.5 μg, 浓度 ≥50ng/μL | cDNA PCR扩增后1D建库 | 需转录本定量:ONT测序数据量 ≥ 15 Gb |
分析内容
- 原始数据质控
- 全长转录本鉴定
- 参考转录组比对
- 基因功能及转录本结构注释
- 差异基因/转录异构体定量
- 功能富集、蛋白互作分析
结果展示
全长转录本识别
建库过程中reads 两端会加上不同的引物,一端为5’,另外一端为3’primer,且3’primer前也存在polyA序列。如果实验中提取的转录本为全长转录本,则完整的插入片段序列就是全长转录本序列。通过判断5’primer、3’primer、polyA的存在以及位置关系,将插入片段序列进行分类,Full-length reads为测序得到的全长序列。
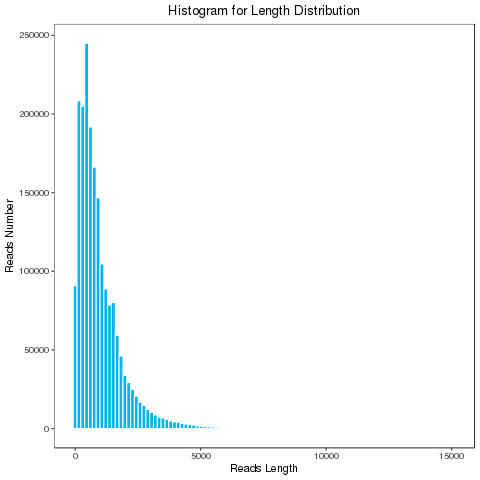 全长序列分布
全长序列分布
融合基因识别
融合基因(fusion genes)是指两个基因的全部或一部分的序列相互融合构成的嵌合基因。其可能是染色体易位、中间缺失或染色体倒置形成的。
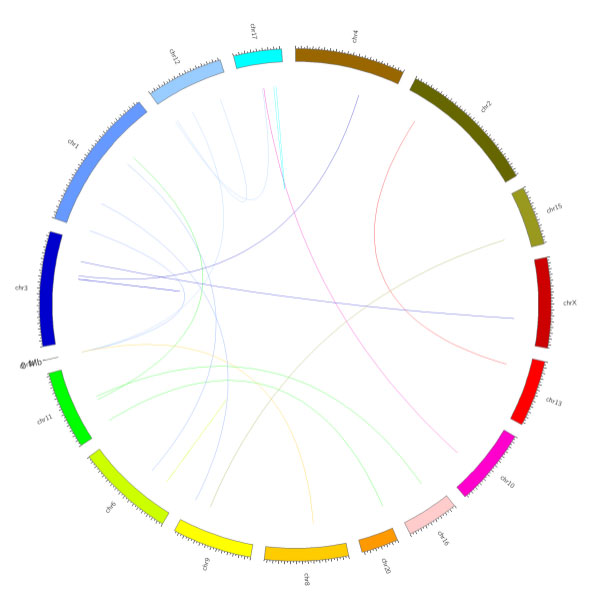 融合基因circos图
融合基因circos图
转录本聚类去冗余
由于同一条转录本在细胞中的一个时间点下可能存在多个拷贝,并且不同的转录本拷贝在测序过程中5’端又可能存在不同程度的降解,所以测序得到的全长reads中,有很多是来源于同一条转录本的冗余序列,需要对全长序列进行聚类去冗余。
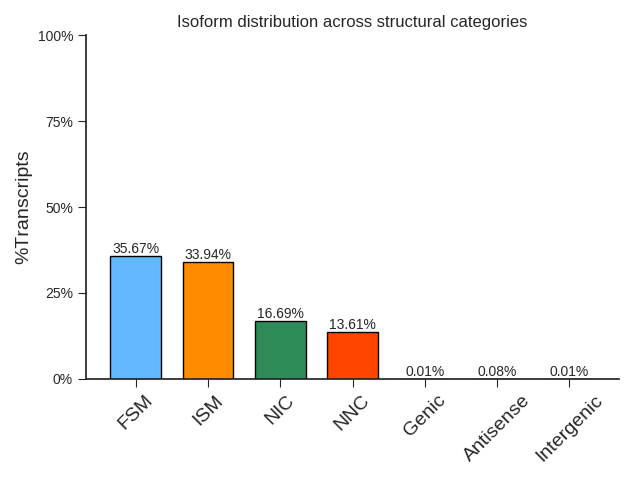 isoform 评估结果
isoform 评估结果
可变剪接识别
可变剪接(Alternative Splice)是调节基因表达和产生蛋白质组多样性的重要机制。
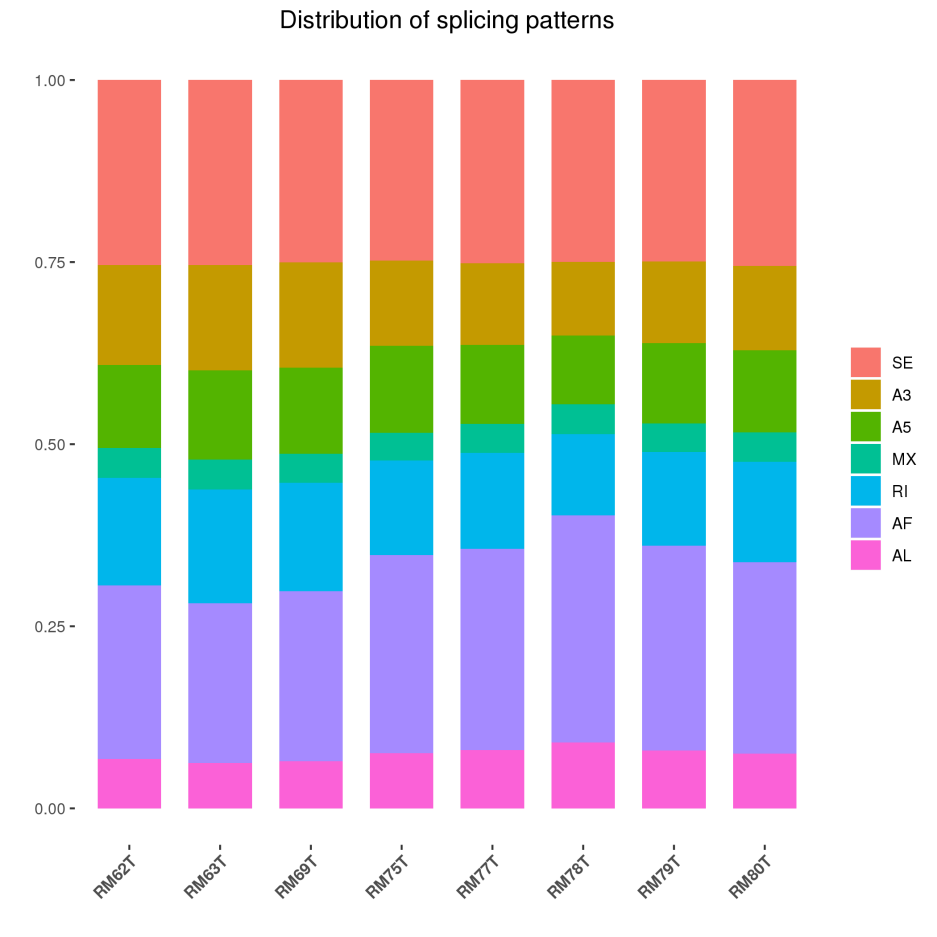 可变剪接类型分布
可变剪接类型分布
差异基因聚类
差异基因聚类分析用于判断不同实验条件下差异基因表达量的聚类模式。
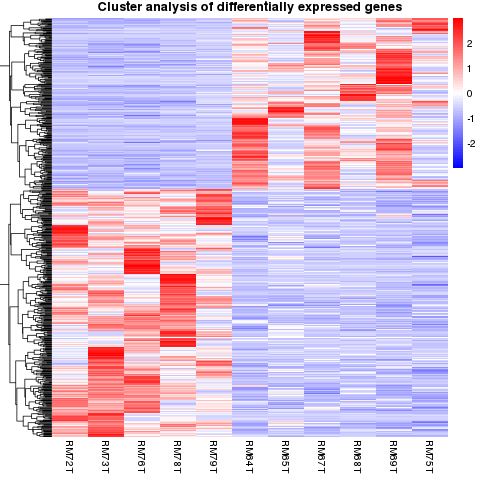 差异基因聚类示意图
差异基因聚类示意图
转录本差异表达
在转录组分析中,DGE(Differential Gene Expression)是指不同条件下基因的差异表达,DTE(Differential Transcript Expression)是指转录本的差异表达(该转录本所属的基因的表达可能发生显著变化,也可能没有显著变化),而DTU(Differential Transcript Usage)是指在不同条件下,基因的表达未发生显著变化,但是属于该基因的转录本的表达有显著变化的情况。
由于Nanopore转录组测序可以在转录本水平进行定量,故而可以进一步分析转录本在不同条件下的差异表达(Differential Transcript Expression)。
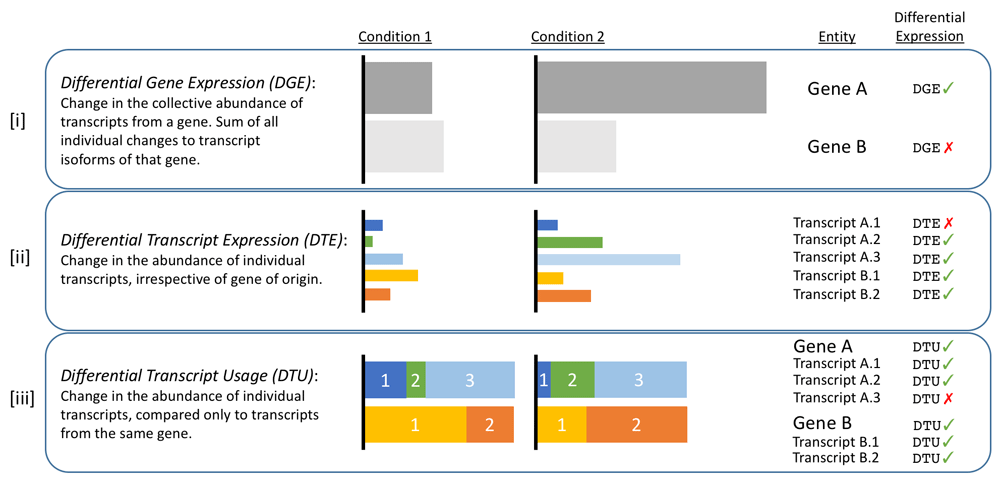 [i]中GeneA在condition1和condition2下发生了差异表达,而GeneB没有;
[i]中GeneA在condition1和condition2下发生了差异表达,而GeneB没有;
[ii]中GeneA的转录本A2和A3发生了差异表达,而A1没有变化,GeneB的转录本B1、B2都发生了差异表达,但是GeneB整体的表达没有变化(此时B1、B2既是DTE也是DTU);
[iii]中GeneA和GeneB的表达在两种condition下没有变化,但是转录本A1、A2和B1、B2的表达有显著变化,属于DTU。
常见问题
1 ONT PCR cDNA相对于PacBio iso-Seq的优势是什么?
在基因水平和转录本水平都可以进行准确的定量,成本更低,以人的样本为例,Sequel II需要2个cell转录本达到饱和,而ONT一个cell可以将6个样本的转录本测到饱和,对样本的需求量也更低。
2 ONT PCR cDNA是否需要做重复?
看研究需求,重点在于转录本的可变剪切,可以不设生物学重复,关心定量分析结果的,一定要做重复,建议至少3个。
案例解析
案例分析一
案例解析
利用ONT全长转录组进行癌症特异性转录变体检测与定量
SF3B1是RNA剪接体关键因子的编码基因,该基因突变会影响RNA正常剪接,导致下游基因表达异常。研究表明SF3B1在多种肿瘤细胞中都出现了较高频率的突变,其中以慢性淋巴性白血病(CLL)患者最为显著。虽然SF3B1突变诱导的选择性剪接模式已经通过RNA-Seq进行了检测,但是短读长限制了在isoform级别上对这一模式的系统鉴定。此外,内含子保留(Intron retentions, IR)在各种癌症中非常普遍,并且可将肿瘤与匹配的正常组织区分开,而短读长技术很容易造成内含子保留事件错误分类,尤其是在复杂的可变剪接区域,并且不能对包含内含子保留的转录本进行定量分析。
该研究利用纳米孔全长转录组数据鉴定并分析了慢性淋巴性白血病样本中SF3B1突变相关的全长转录本。纳米孔测序技术能够对全长转录本进行测序,直接获得全长isoform,并准确识别内含子保留,同时高通量的PromethION测序仪能够产生足够数量的全长序列转录本用于癌症特异性转录变体检测和定量。
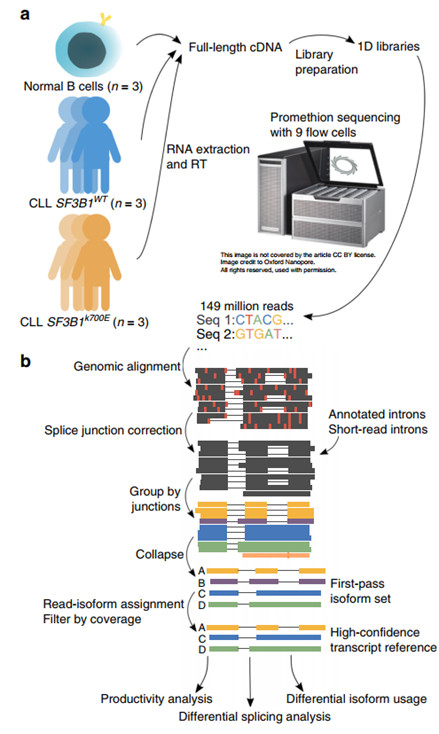 图1 长读长纳米孔测序和FLAIR序列分析流程
图1 长读长纳米孔测序和FLAIR序列分析流程
研究者分别选取3个SF3B1无突变CLL患者样本(CLL SF3B1WT)、3个SF3B1K700E突变CLL患者样本(CLL SF3B1K700E)以及3个普通B淋巴细胞样本进行ONT全长转录组测序,共获得约257M条reads(图1a)。随后,研究者开发了FLAIR流程用于识别高可信转录本,该流程包括三个主要步骤:比对,校正和整合。首先将所有样本的原始reads与基因组进行比对,识别一般的转录本结构,随后利用短读长测序数据纠正reads中因测序错误、碱基缺失和对齐困难导致的错误剪接点,然后将具有相同剪接点顺序的reads整合到对应的isoform组,再将所有reads重新比对至这些isoform上,最后将少于三条reads支持的isoform过滤掉,剩下的具有足够覆盖度的isoform构成最终的高置信度isoform集合(图1b)。
利用FLAIR,研究者共鉴定出326,699个高可信度剪接isoform,其中大多数(90.0%)为未注释的isoform。这些未注释的isoform中有142,971个是已注释的剪接连接形成的新组合,其余的是包含内含子保留(21,700)或新的外显子(3,594)的isoform。研究者利用纳米孔数据中验证了前人报导的与SF3B1突变相关的3’剪接位点的差异变化。结果表明,长reads不仅使能够识别突变的SF3B1改变的剪接位点,同时还可将这些剪接位点异常事件及其他类似事件与对应的全长isoform相关联。
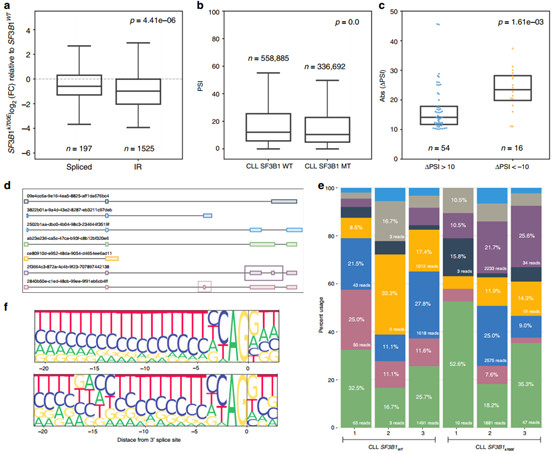 图2 内含子保留事件在CLL SF3B1K700E中被更强烈地下调
图2 内含子保留事件在CLL SF3B1K700E中被更强烈地下调
随后,研究者探索了与SF3B1突变相关内含子保留isoform变化,通过比较CLL样品间的表达谱差异,发现SF3B1K700E样品中的内含子保留isoform表达较SF3B1WT样品中普遍下调(图2a)。进一步研究发现,利用重新分析突变SF3B1的CLL、Nalm-6细胞系和TCGA BRCA短读长数据集证实了这种下调(图2d-e)。
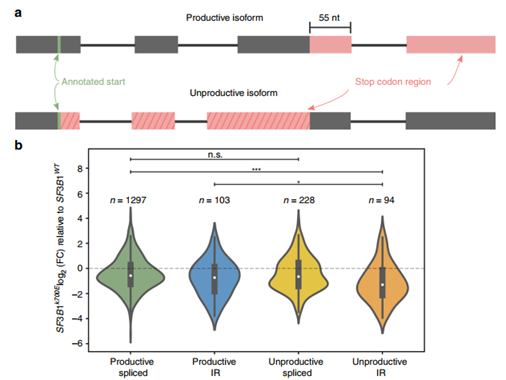 图3 突变的SF3B1下调了“无效的”内含子保留的转录本
图3 突变的SF3B1下调了“无效的”内含子保留的转录本
此外,研究还发现全长reads不但可以提升内含子保留转录本的识别,而且能够更好地估计有效和无效isoform的丰度(图3a)。研究者将SF3B1K700E和SF3B1WT样本之间差异表达的isoform,按照是否有效、是否包含内含子保留进行分类,结果表明无效的、内含子保留的转录本表达下调(图3b),GO分析发现这些无效的、内含子保留的转录本与激酶信号通路相关。研究者推测它们可能是滞留在细胞核中的内含子,表达下调可能导致激酶相关基因的表达,从而支持肿瘤的增殖,但仍需要进一步的实验验证。
参考文献:Tang, A.D.,Soulette, C.M., van Baren, M.J. etal. Full-length transcript characterization of SF3B1 mutation in chroniclymphocytic leukemia reveals downregulation of retained introns. Nat Commun 11, 1438 (2020). https://doi.org/10.1038/s41467-020-15171-6
官方微信公众号

希望组

希望组科技服务

希望组诊断服务