Science报道丨西湖大学俞晓春团队解析迄今最完整小鼠基因组图谱
对于生命而言,DNA的重要性不言而喻。它既支撑生命的构造和性能,也储存着个体生长、孕育、凋亡“从生到死”的全部相关信息。正因如此,着眼于健康与疾病的谜题,人类不仅需要翻开、阅读这本生命之书,也亟需“读完”它——
北京时间2024年12月6日凌晨,《科学》(Science)杂志在线发表了西湖大学生命科学学院、西湖实验室俞晓春团队最新成果“完整的端粒到端粒小鼠参考基因组序列(The complete telomere-to-telomere sequence of a mouse genome)”,报道了该团队在解析小鼠参考基因组方面取得的重要突破。这意味着人类历史上第一次看清小鼠基因组DNA全貌。
论文截图
原文链接:
https://www.science.org/doi/10.1126/science.adq8191
现在,请用上一些想象力,一起走入基因组DNA的殿堂,造访大自然塑造的“生命密码”。
想象你的面前出现了一座汗牛充栋的图书馆,这是隶属于某个人类同胞的一个细胞核。你步入其中,看到了几十排标注着“染色体”的书架。你随机选了一个架子,抽出了几本书,发现书的封面上都写着“DNA”。接着,你翻到其中一本的目录页,上面指示了“本书共含有X个基因”。你随意浏览了不同基因的章节,意识到这些篇章仅由四个字母构成——A、T、C、G——这些叫作“碱基”的字母不断变换顺序、排列组合,最终写完了全书……
很好,现在你已经了解了基因组DNA的基本面貌。
正如开头所述,如果我们想获得一个生命体的所有遗传信息,就需要知晓全部基因组DNA的情况,这意味着要阅读完所有染色体“书架”上的DNA之书,知道这些书的每一个字母,即A/T/C/G是如何排列的。关注生物体所有DNA(即整个基因组)的科学,就是基因组学。迄今,基因组学领域的一个重要研究目标,正是获得完整的、精确的基因组序列,这对于我们理解基因组的结构和功能至关重要。
事实上,读取这些碱基字母排序的过程,正是“大名鼎鼎”的基因组DNA测序。
1977年,弗雷德里克·桑格发明了第一代测序技术,特点是只能测试一个基因的某个部分,最多一个基因。本世纪初,第二代测序技术问世,它克服了前一代的缺点,一次能读取成千上万的短DNA片段,因此也被称作高通量测序技术;但它依然存在症结:能读取的DNA片段过短,通常在100-300个碱基对(bp)之间。2010年左右,第三代测序技术诞生,实现了对每一条DNA分子的单独测序;换句话说,现在我们能够读取较长的DNA片段了,可以达到10-50千碱基对(kb,1kb=1000bp)甚至更长。
由于人类基因组包含大约30亿个碱基对,能够读取更长片段的第三代基因测序技术的出现,为科学家破解完整的人类基因图谱的进程按下加速键。2022年3月31日,《科学》发表文章报道了名为“端粒到端粒联盟”的国际科学团队,完成了第一个完整的、无间隙的人类基因组序列,填补了2003年“人类基因组计划”遗留下的8%尚未读取的基因区域。
在大洋彼岸的中国浙江杭州的西湖大学,俞晓春实验室当时的博后、现在的助理研究员李麒麟及时关注到了这条新闻。这令这个团队感到无比振奋,因为他们日常“打交道”的小鼠身上,正存在相似的瓶颈。目前小鼠的基因“档案”中,最完整的是参考基因组GRCm39,同样也存在约7~8%未被解析的区域。
西湖大学生命科学学院科研副院长、西湖实验室科研副主任俞晓春教授长期致力于DNA损伤修复机制和癌症发生发展的研究;简单来说,就是DNA受损引发的癌症的诊断、检测与治疗。而小鼠,是生命科学研究中最常见的实验动物和模式生物,这是因为许多生物实验不宜在人体内进行,因此,小鼠的基因组DNA信息直接关系到人类健康的探索。也正因如此,人类对小鼠基因组DNA的认知与这个团队的研究密切相关。
既然人类的“基因拼图”已完成,想必小鼠的“拼图”也胜利在望了?令他们没想到的是,这一等就是一年。
亲自做基因测序,对俞晓春实验室来说,实属一个“无心插柳柳成荫”的课题:直到2023年4月,他们都在等待两家资金雄厚、早已对外宣布下场的美国与英国科研机构做完并发布小鼠的完整基因组DNA图谱。
为什么他们如此关心小鼠这尚缺的7%-8%序列?这是因为,这些未知的基因组DNA里或许隐藏着一些至今无法解释的遗传性疾病的谜底。
这些“空白”尤其存在于异染色质和核糖体DNA(rDNA)区域。这些区域富含重复的基因序列,即一些反复出现的,看似近乎一模一样、但实则有细微区别的片段——你可以想象为许多块极其相似的拼图。二代基因测序技术仅能测出其中的一段(且由二代技术完成的小鼠基因组图谱中还有错误),对完整的排序序列“束手无策”;而三代技术可以“完全看清”。
时至2023年的春天,迟迟不见欧美的实验室发布“大新闻”,这个实验室最终决定自己动手拼完这幅小鼠基因组“拼图”。“(全球)剩下的人一直在等,但我们不想等了。”俞晓春回忆说。
这个诞生于意外的课题,研究过程相当顺利,历时一年就完成了。
简单来讲,俞晓春团队综合了众多三代基因测序技术,让它们互相补足,开发了一把能够充分挖掘小鼠基因的“金铲子”。他们以最常用的小鼠C57BL/6的单倍体胚胎干细胞(mhaESC)为样本,进行了基因测序和组装,获得了长度为2.77 Gbp(表示十亿个碱基对)的完整的高质量小鼠参考基因组序列,其中包含215.23 Mbp(表示一百万个碱基对)先前未被鉴定的序列,填补了约7.7%的基因组空白。

mhaESC基因组与先前参考基因组的共线性比对结果
如果你对他们基因组DNA “拼图”的步骤感兴趣,这个流程大致是这样的:第一步,测序技术把所有拼图(即片段)上的图案(即碱基对)读完;接着,计算机对这些信息进行数据处理;最后,复杂算法会完成“拼装”(即基因组组装),形成完整的全貌。这个过程涉及到了PacBio HiFi、Oxford Nanopore超长、Illumina短读长、Hi-C和BioNano光学图谱等多项基因测序技术。
那么,这些研究人员具体取得了哪些关于小鼠基因的新发现呢?
首先,发现了新的蛋白质编码基因。顾名思义,这些基因的作用是编码对应的蛋白质。与先前的参考基因组版本相比,本研究额外注释了639个蛋白质编码基因,其中先前未被发现的全新的蛋白质编码基因有140个(这是因为639个基因中部分为已知基因的“重复”拷贝)。这些新的蛋白质编码基因可能参与多种生物学过程,为未来的研究提供了新的方向。
第二,较精确地“看清”核糖体DNA的基因序列。核糖体是细胞内的“蛋白质工厂”,负责合成蛋白质。核糖体DNA是细胞中的一种特殊DNA,它专门负责编码核糖体的RNA(rRNA)——一种核糖体的重要组成部分,帮助核糖体合成蛋白。用简洁的比拟来说,核糖体DNA给出了细胞内rRNA的“蓝图”。这个发现为进一步解析核糖体潜在的蛋白质翻译功能的差异性提供参考。
第三,解析了着丝粒区域的基因序列详情。着丝粒是染色体上的一个特殊区域,帮助染色体在细胞分裂时,将遗传物质平均分配到两个新的细胞中。本研究的结果显示,小鼠各染色体之间的着丝粒长度具有明显差异,且序列内部富含转座元件和片段重复(SD),同时还有散在的基因分布,表明该区域可能会进行活跃的转录和转座事件,驱动着丝粒区域进行适应性改变等行为。对着丝粒区域的解析,有助于理解因着丝粒功能缺陷导致的染色体重排、非整倍性等相关疾病的发病机制。
让我们总结一下。从科学意义上来说,俞晓春实验室的这项研究,通过综合“长读长”第三代测序技术成功完成了小鼠基因组的端粒到端粒组装,填补了现有参考基因组中的空白区域,揭示了新的基因和结构变异,“拼完”了小鼠基因组图谱的“拼图”。这些发现不仅提高了对小鼠基因组结构和功能的理解,也为基因组学研究提供了重要的技术参考和数据资源。
在这项研究中,两位一作作者,分别发挥了科研所长,刘俊丽助理研究员负责湿实验及论文图片,李麒麟助理研究员负责干实验及文稿;通讯作者俞晓春教授负责“掌舵”课题的大方向以及论文的完善。
“你们在研究过程中遇到最大的难点是什么?”这个问题竟然有朝一日成为了实验室“答不上来”的问题。正如前文所言,这个课题进展势如破竹,投稿过程也十分顺利。
但要在科研的疆域取得成果,并非一日之功。这项研究的顺利开展,既得益于俞晓春自在美国密歇根大学医学院内科系成为独立PI后,对染色体近20年的研究积累;同时,也与两位一作作者历经过的、作为一名科研工作者的磨炼与自我调整息息相关。
刘俊丽,是西湖实验室第一批“开拓学者”之一,曾在科研的路途上迷茫过、也曾经历过gap的时光,但她最终选择加入俞晓春实验室,尽管那意味着要完全改变研究方向,需要从“0”开始。如今,她分享说:“做科研,任何一个方向都有研究意义。我觉得实验取得的任何结果都能带给我快乐,这是为什么我要坚持做科研的原因。”
如果说这个课题有一个发起人,那非李麒麟莫属:他是俞晓春团队第一个注意到人类基因组序列完成的人。出于对遗传学和基因组学的兴趣,他从大学本科直至在美国做博后阶段都专注于生物信息学。李麒麟说:“但我发现做纯数据并不能对实际情况有很好的了解,所以最后我选择了俞老师的实验室,这里有湿实验的实时结果给出反馈,这样我再去做数据分析,研究能更好地开展。”
当然,俞晓春实验室剑指的始终并不是小鼠基因组真容本身,而是希望利用这把“基因组之铲”探索遗传性癌症、发育性疾病未解的致病机理。“支线”的故事已完成,接下来,让我们一起静待这个实验室的“主线”诞生更多助力人类攻克顽疾的成果。
西湖实验室助理研究员刘俊丽博士和李麒麟博士为本文的共同第一作者,西湖大学生命科学学院科研副院长、西湖实验室科研副主任俞晓春教授为通讯作者。本研究得到国家自然科学基金、浙江省自然科学基金、浙江省“尖兵”&“领雁”项目、杭州市领军型创新创业团队、西湖教育基金会和西湖实验室提供的经费支持,同时感谢西湖大学生物医学实验技术中心、实验动物中心及高性能计算中心等平台的支持。

 图 1 百合基因组和多组学分析
图 1 百合基因组和多组学分析





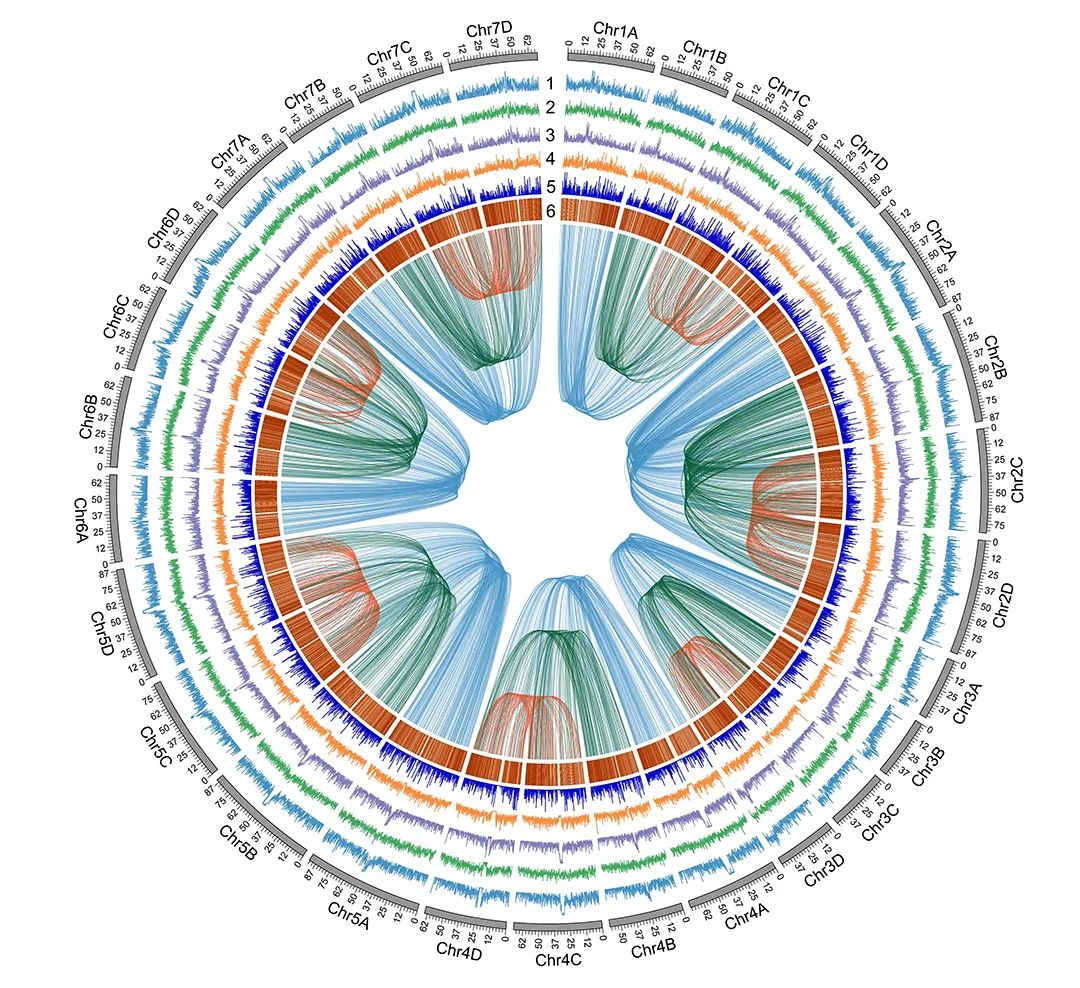



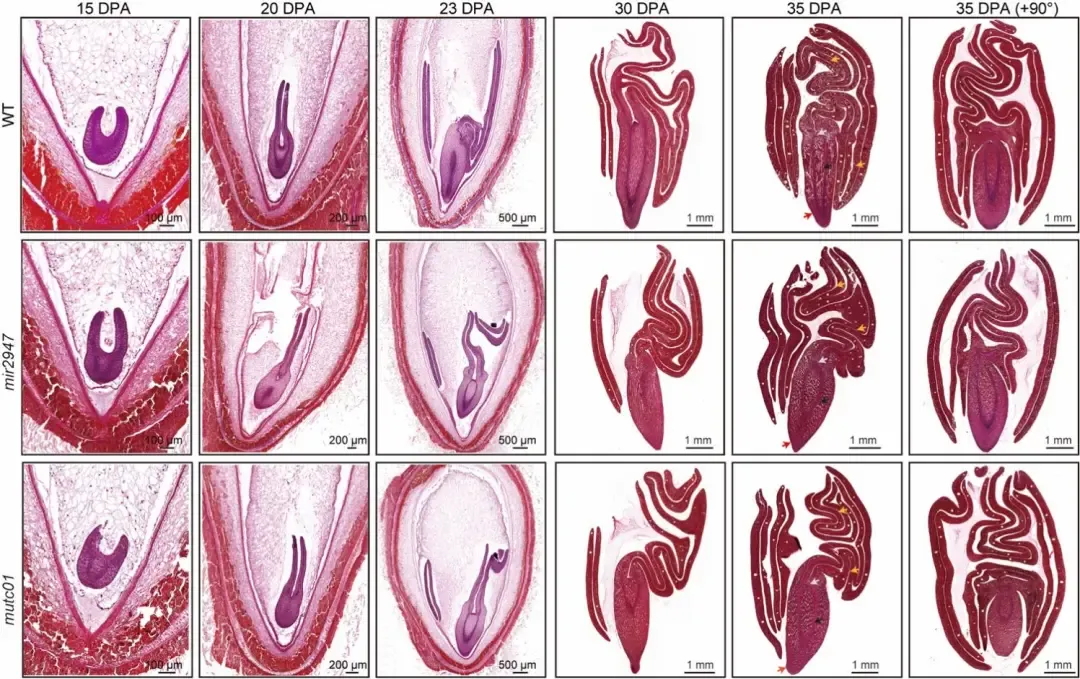
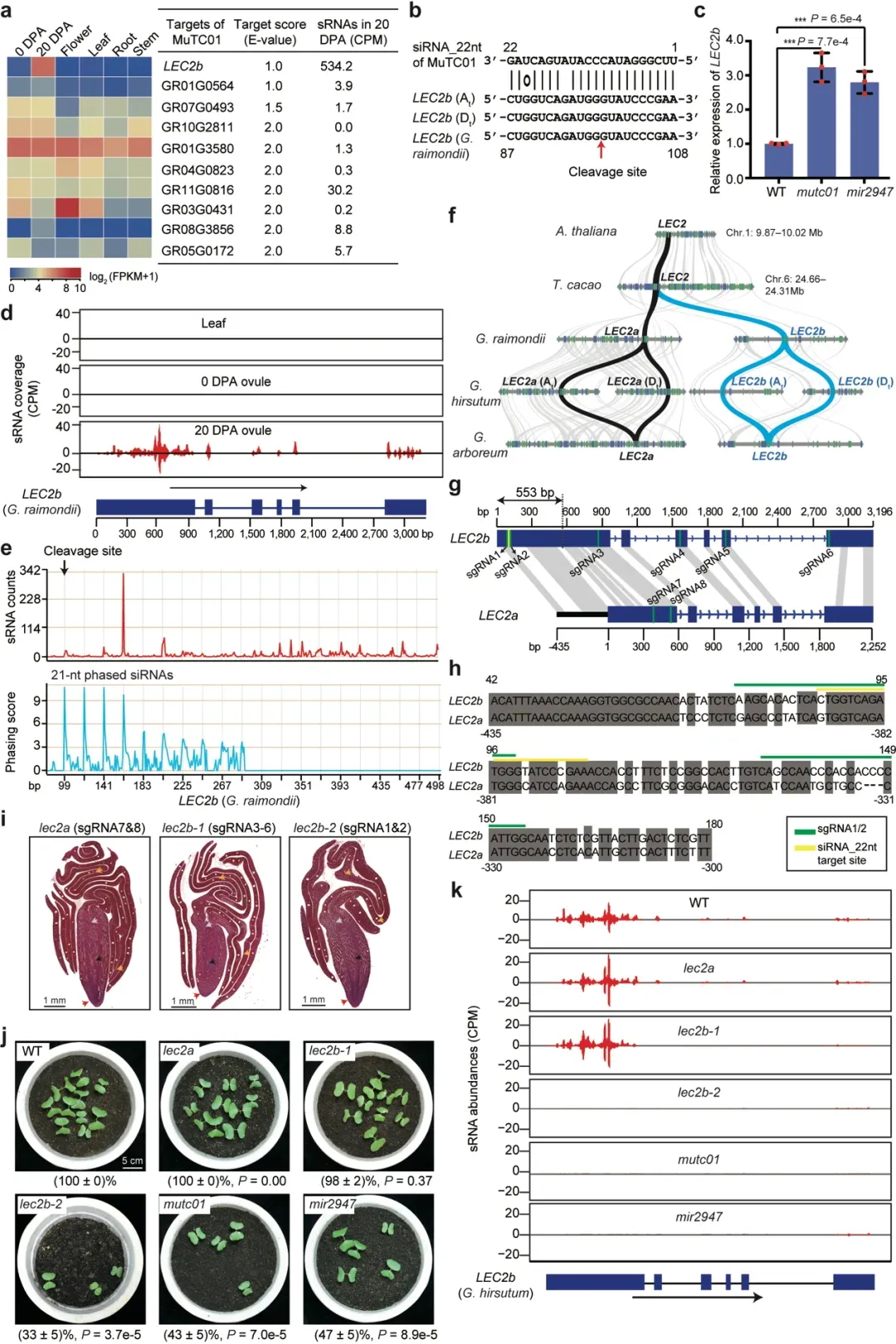
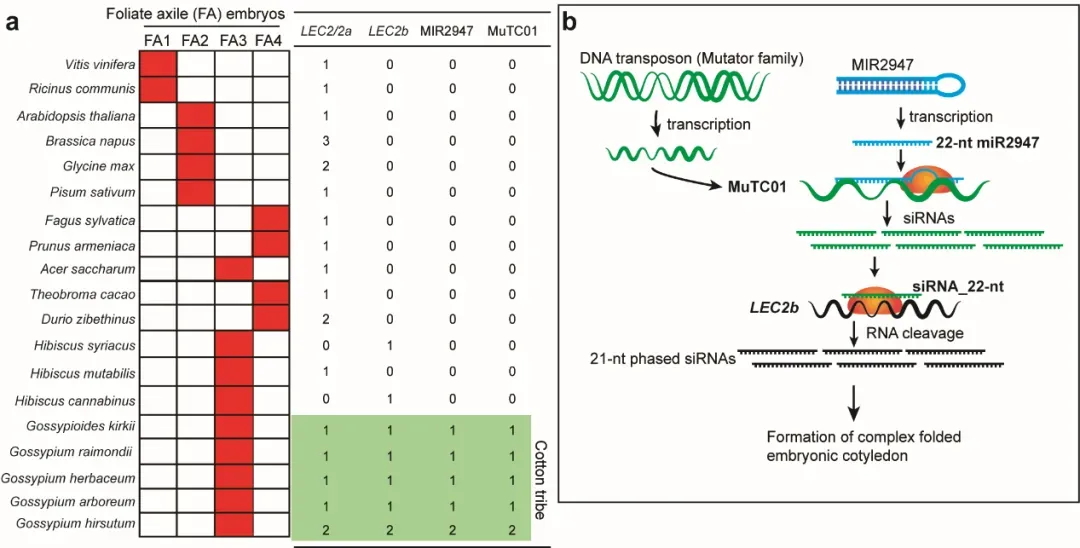

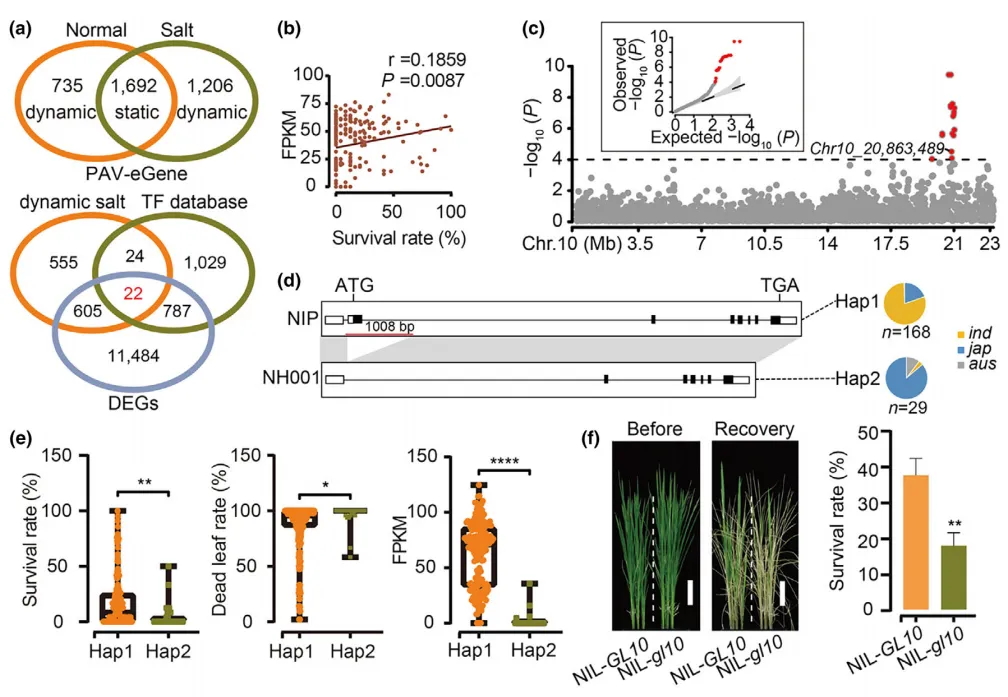
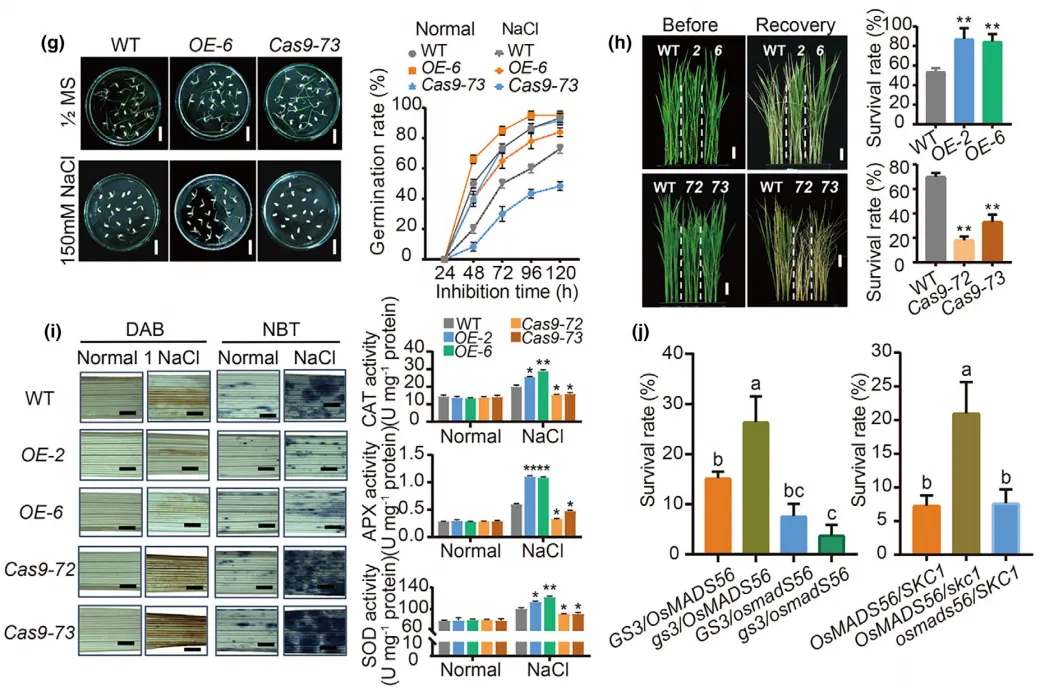

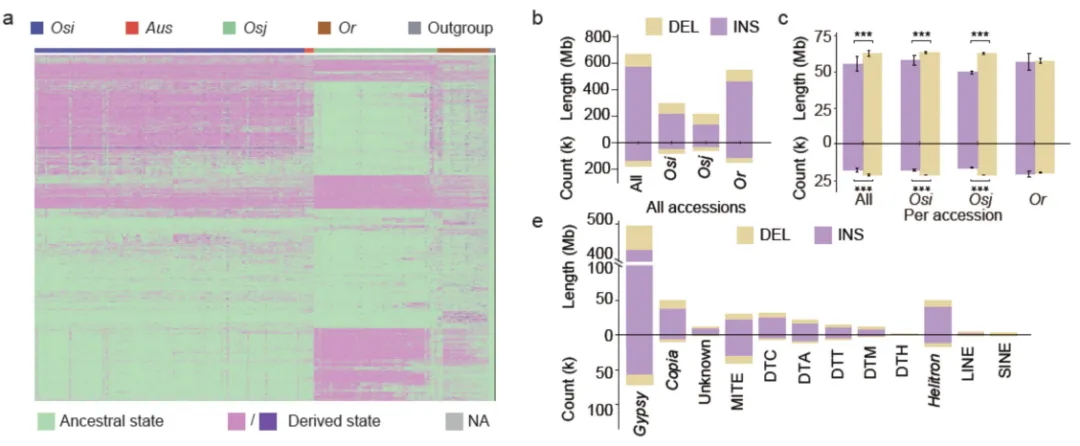
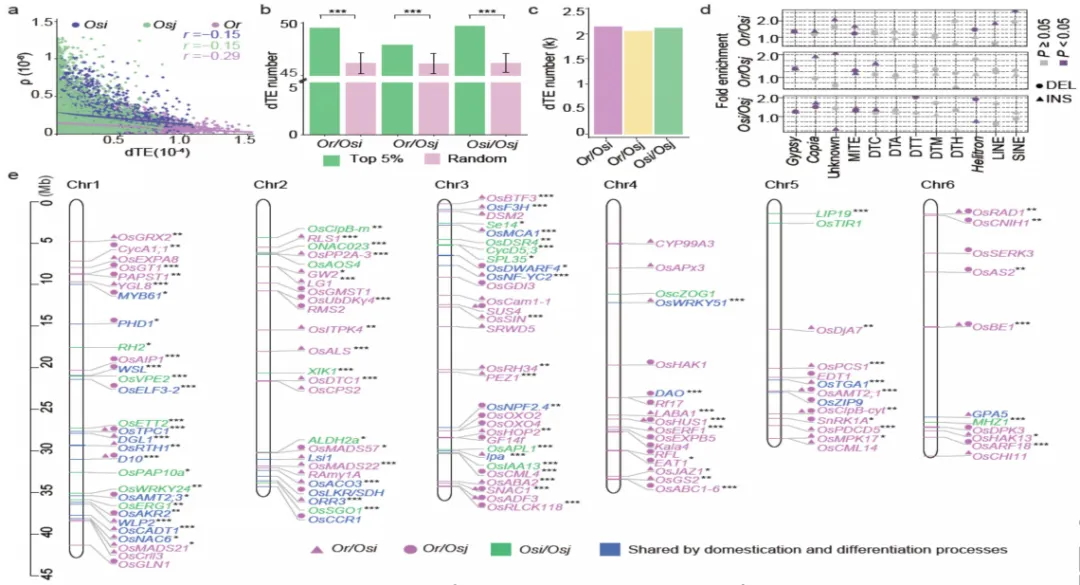
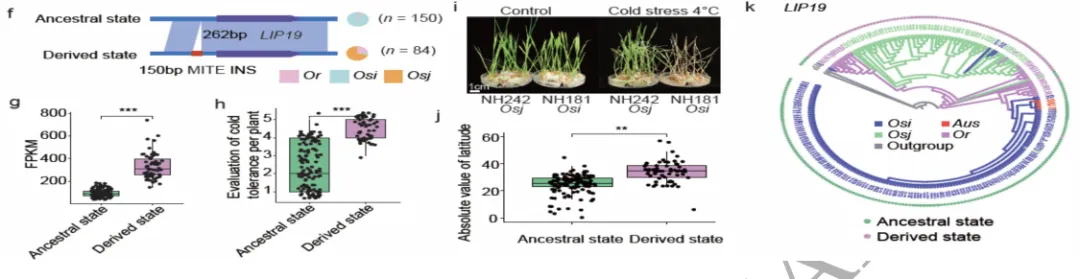
 图3 TE变异影响水稻基因表达和农艺性状
图3 TE变异影响水稻基因表达和农艺性状