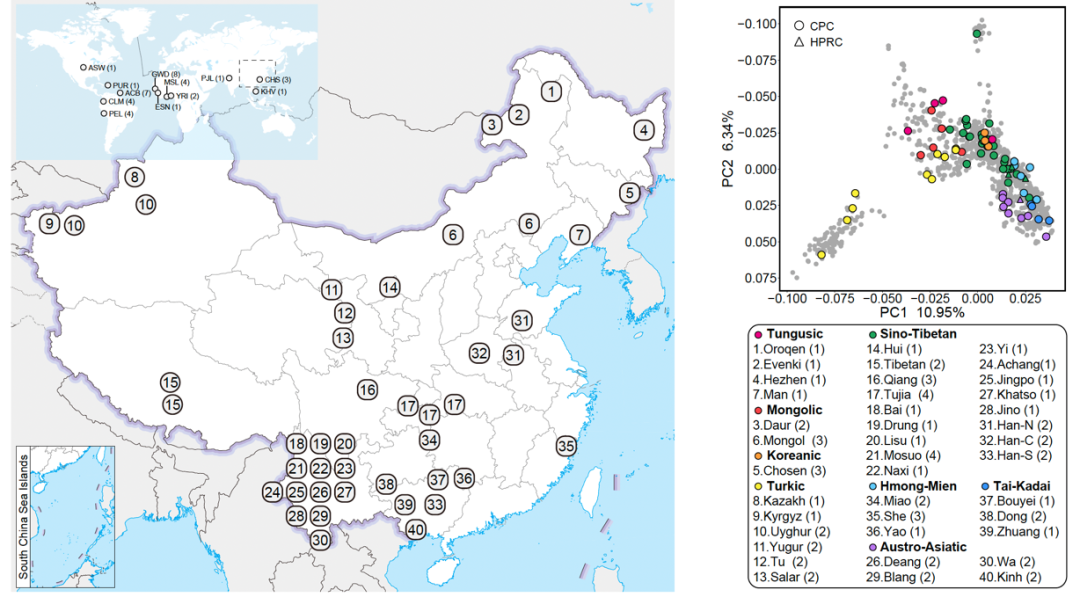
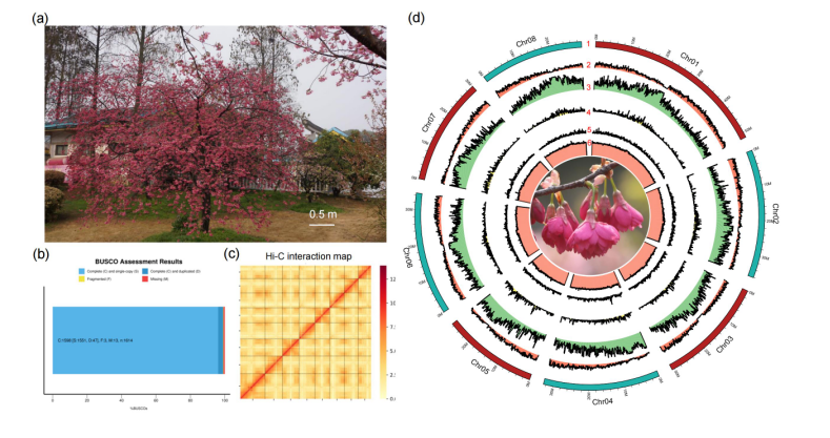
希望组参与全球首套多组学标准物质“中华家系1号”的最新研究成果!
生物医学研究已经步入大数据和大科学时代。一方面,多组学数据分析已成为生命科学前沿领域最重要的研究工具之一,多维度数据挖掘与整合分析,可以帮助科学家实现从基因组到表型组、贯穿微观和宏观尺度的系统分析,极大提高了人类解读复杂生命系统的能力,对更加深刻、精准地破解肿瘤、遗传病等各类疾病的发病原因与微观机制,寻找更有效的干预手段奠定了重要基础。另一方面,要破解人类健康、生命起源等重大科学问题,需要进行全球合作,开展分布式的国际大科学计划。然而,没有高质量的数据生成、高可靠的数据分析与整合以及全球科学界一致认可的统一标准,多组学数据分析研究就失去了稳固的“地基”,全球范围的生命科学国际大科学计划也将无从谈起。如何解决类似的难题?研发国际科学界广泛认可的多组学标准物质至关重要。
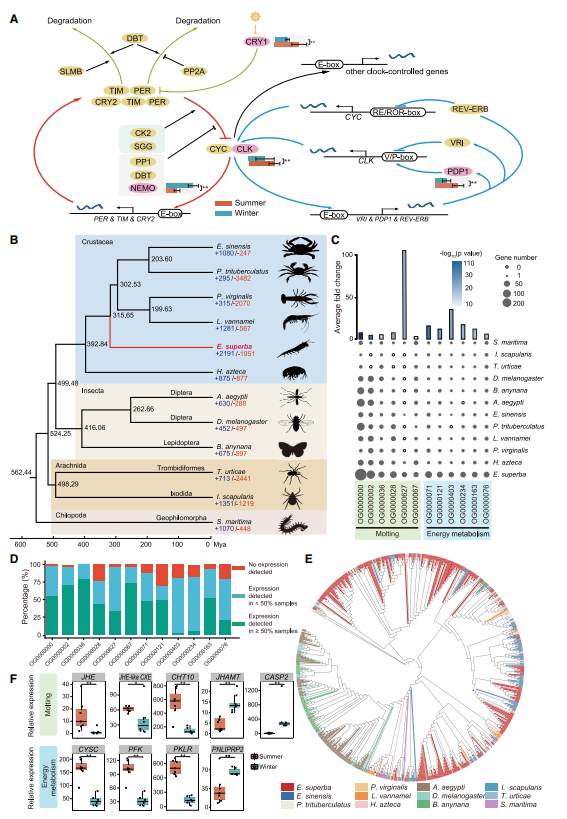
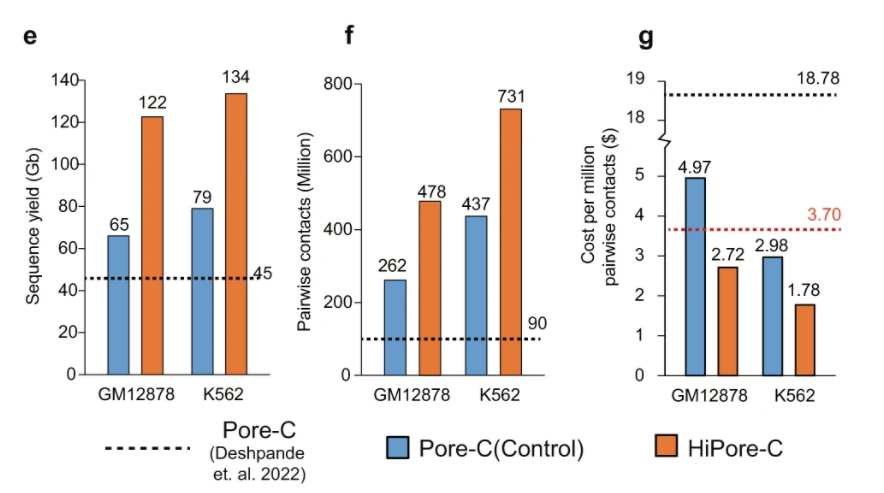
北京时间2023年9月7日晚,国际学术期刊《自然·生物技术》(Nature Biotechnology)在线发表了由复旦大学/上海国际人类表型组研究院石乐明、郑媛婷团队联合中国计量科学研究院方向、董莲华团队,国家卫健委临床检验中心李金明、张瑞团队共同研发的全球首套多组学标准物质“中华家系1号”的最新研究成果。同期刊发的2篇科研论文分别聚焦:“使用基于中华家系1号标准物质的相对定量进行多组学数据整合(Multi-omics data integration using ratio-based quantitative profiling with Quartet reference materials)”和“中华家系1号 RNA标准物质与基于比值的分析方法提高了转录组数据的质量(Quartet RNA reference materials and ratio-based profiling for assessing and improving the quality of transcriptomic data)”。这也标志着中国科学界自主研制、获批为“国家一级标准物质”的“中华家系1号(Quartet)”多组学标准物质的研发和效用得到了国际同行的认可,开创了生物医学“度量衡”新体系,将提升生命科学创新的源头质量,为全球推进人类表型组计划奠定坚实的标准基础。
标准物质是高质量生物医学创新研究的“标尺”与“砝码”
在生命科学研究中,针对相同研究样本在不同平台、不同实验室、不同批次所产生的组学数据往往存在“批次效应”,导致不可重复数据和错误结论,严重影响科研结果的可信度与质量。而现实生活中,类似“批次效应”的危害更大:在临床检验中,同一个指标在不同的医院检验结果会出现差别,这种数据差别一旦过大甚至会导致错误的临床治疗决策,耽误疾病的预防和诊治。
要解决批次效应这一影响生命科学与生物医学多组学研究源头质量的“拦路虎”,就必须研发相应的标准物质。标准物质是指具有足够均匀性和稳定特性的物质,可作为生物分析研究的“标尺”与“砝码”。在生物医学研究中,标准物质可用于评估不同实验室、不同平台、不同批次的数据质量,有助于排除实验条件和技术差异带来的误差,确保数据的一致性和可靠性。而多组学研究的普及,亟需科学界研发多组学标准物质。
统一的标准是生命科学领域国际大科学计划全面推进的关键基础
由于测量和研究的对象涉及到人类自身,因此生命科学领域的大科学计划与其他学科领域存在显著差别。分布式,即在不同大洲和国家各自实施,而不是集中式地开展研究是生命科学领域国际大科学计划的主要组织模式。这就对相关大科学计划在科研和实施过程中所参照的标准和质量控制提出了极高的要求。基于公认的基准——标准物质,统一相关研究的测量标准和数据标准,使得全球不同实验室针对同一类研究的数据可以参比,是生命科学领域能够实质性开展大科学计划的重要前提和基础。
作为人类基因组计划之后,生命科学领域的下一个战略制高点和重大科学计划,人类表型组计划在规划之初就把研发标准物质和统一全球科研标准作为重中之重。在国家和上海市支持下,中国相关科研团队在人类表型组的精密测量、标准物质研发、质量控制、数据处理等各个方面在全球范围内率先开发和制定相关SOPs、标准和质控体系,并通过国际和中国两大协作组网络,推动协同全球不同地区的实验室在同一标准下开展表型测量与研究。
相关团队已经完成了对2万余种表型开展测量的质控标准研发与SOP编制工作。2021年10月,由石乐明教授牵头起草的国际标准ISO/TS 22690:2021 《基因组信息学 高通量基因表达数据可靠性评估》(Genomics informatics—Reliability assessment criteria for high throughput gene—expression data)发布。该标准规定了高通量基因表达数据的可靠性评估标准,适用于基因芯片、新一代测序的基因表达数据的准确性、复现性、可比性的评估应用。同年10月,在上海市市场监督管理局的指导下,“上海市标准化创新中心(国际人类表型组)”获批成立,成为上海市首批6家新型标准化技术组织单位之一,正在全面引领国内外人类表型组标准化研究与创新。
此次“中华家系1号”多组学标准物质最新研究成果的国际发表,是中国科学家引领人类表型组计划实质性推进所作出的又一里程碑式的贡献。可以说,在人类表型组科研质量控制与标准体系构建中取得的一系列先发优势,进一步奠定了中国科学界在人类表型组计划中的引领地位。希望组为本研究提供三代测序和分析服务。
“二十年磨一剑”,打造全球首个多组学标准物质
在“中华家系1号”研发成功之前,全球尚无任何一种生物学标准物质能够具备多组学研究需要的特性。作为全球首套多组学标准物质,“中华家系1号”涵盖了同一来源样本的多种分子水平的特性,如DNA、RNA、蛋白质、代谢物等。这些标准物质的引入为生物医学研究和临床应用提供了可信赖的计量标准,为高质量、高可靠性的多组学研究提供了坚实基准。
“中华家系1号”多组学标准物质,源自复旦大学领导建设的泰州大型人群队列中的一个同卵双胞胎家庭的永生化B淋巴母细胞系。“中华家系1号”是国际上首套包括DNA、RNA、蛋白质、代谢物在内的多组学标准物质,旨在确保分子表型组数据跨批次、跨实验室、跨平台、跨组学的可比性和准确性。其中,DNA、RNA标准物质已经获得了国家市场监督管理总局颁发的8项国家一级标准物质证书(GBW 099000-GBW 099007),是我国首次获批的组学标准物质,在生命科学领域开创了一种全新的标准物质研制模式。

图1:“中华家系1号”(Quartet)多组学标准物质


图2:国家一级标准物质证书(GBW 099000-GBW 099007)
在“中华家系1号”的研制过程中,研究团队通过在国内32个研究中心运用24种主流技术平台对标准物质进行了深入全面的表征,获得了包括基因组、表观基因组、转录组、蛋白组和代谢组在内的多组学大数据。在此基础上,研究团队提出了一系列质量控制指标,构建了高置信的标准数据集,为多组学技术、实验室性能、分析算法的评估提供了高质量的“基准真值”。
据悉,基于“中华家系1号”DNA和RNA标准物质,国家卫生健康委临床检验中心已于2021年和2022年分别开展了全外显子测序和转录组测序的全国科研与临床实验室的室间质评研究,参加单位超过100 家,并将逐步开展表观基因组、蛋白质组、代谢组等多组学室间质评,以促进我国科研和临床实验室多组学检测数据质量的不断提升。
据石乐明教授、郑媛婷副教授介绍,在严格遵守我国人类遗传资源管理条例并获得国家批准的基础上,上海国际人类表型组研究院和复旦大学大力推动“中华家系1号”多组学标准物质走向全球,已经在国内外100多家单位进行了广泛应用,扩大了中国标准物质的国际影响力。例如,欧洲转化医学研究先进基础设施(European Advanced Translational Research Infrastructure in Medicine (EATRIS) Plus)已经采用“中华家系1号”多组学标准物质对EATRIS-Plus联盟的多家单位在多组学数据产生和数据分析方面的性能进行客观评估。欧方正与上海国际人类表型组研究院等中国代表性机构共同探索、积极推动构建多组学生物数据质量的国际标准。
基于多组学标准物质的质量控制将保证生物医学创新源头的高质量
未来的生物医学研究中,多组学分析是一个贯穿基因型到表型的整合过程,从数据生成和数据整合程序的每个环节都会影响最终结果。因此,必须对每种组学数据从样品到结果的完整流程进行全面能力验证和质量控制。
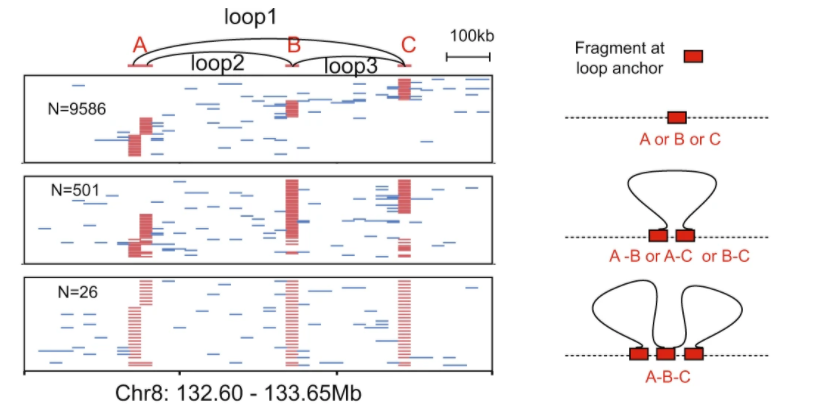
本次发表的最新成果证明:“中华家系1号”不仅具有天然的家系关系,样本之间微小的内在生物学差异可为数据整合提供高灵敏度的可靠性评估。此外,这些基于同一来源细胞系制备的多组学标准物质包含了从DNA到RNA再到蛋白质的信息流,遵循中心法则,可用于验证整合结果是否反映跨组学分子间的逻辑关系。
在传统的基于组学标准物质的质量控制中,通常将标准数据集视为“金标准”。然而,这些数据集只能评估高置信基因组区域中的变异和稳定检出的高表达分子特征,并且受到构建时采用的技术平台和分析方法的限制,不适用于对新技术的质量评估。本研究提出了不依赖标准数据集而仅基于家系个体间生物学关系的质量评估参数:对于定量组学数据,信噪比(Signal-to-Noise Ratio,SNR)可用于评估测量系统能否识别不同样本组之间的固有生物学差异,这是转录组等定量组学分析的基本目标;对于定性组学数据,同卵双胞胎之间胚系变异的一致率和家系个体间孟德尔符合率,可以实现在全基因组范围内对变异检测准确性的客观、无偏好的质量评估。通过与标准数据集的联合使用,多组学数据的质量控制体系更加完善,为各类新兴技术的质量评估提供了可能。希望组为本研究提供三代测序和分析服务。
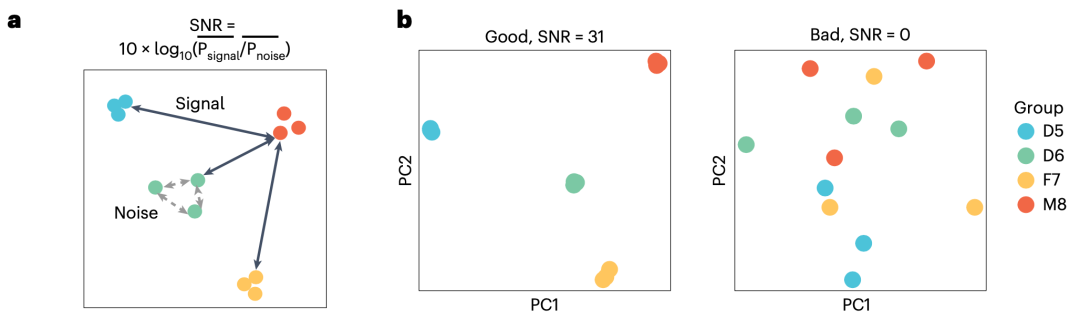
图3:信噪比(SNR)
本次的研究成果最终提出了多组学分析的质量控制指标和整合的最佳实践建议:
定量组学分析需从“绝对”定量向“相对”定量转变,各批次使用固定的标准物质可有效控制批次效应;
多组学整合结果的质量可以结合家系信息、中心法则进行评估,如样本分类、跨组学特征关系识别的准确性等。
多组学分析在生物医学研究中具有广泛的应用前景,为了确保结果的准确、可靠、可重复,研究人员需要遵循质量控制和最佳实践建议。这一研究为多组学领域的规范化、标准化发展奠定了坚实基础,指明了提高多组学分析质量和可信度的重要途径,对促进多组学研究的高水平、高质量发展具有重要意义。
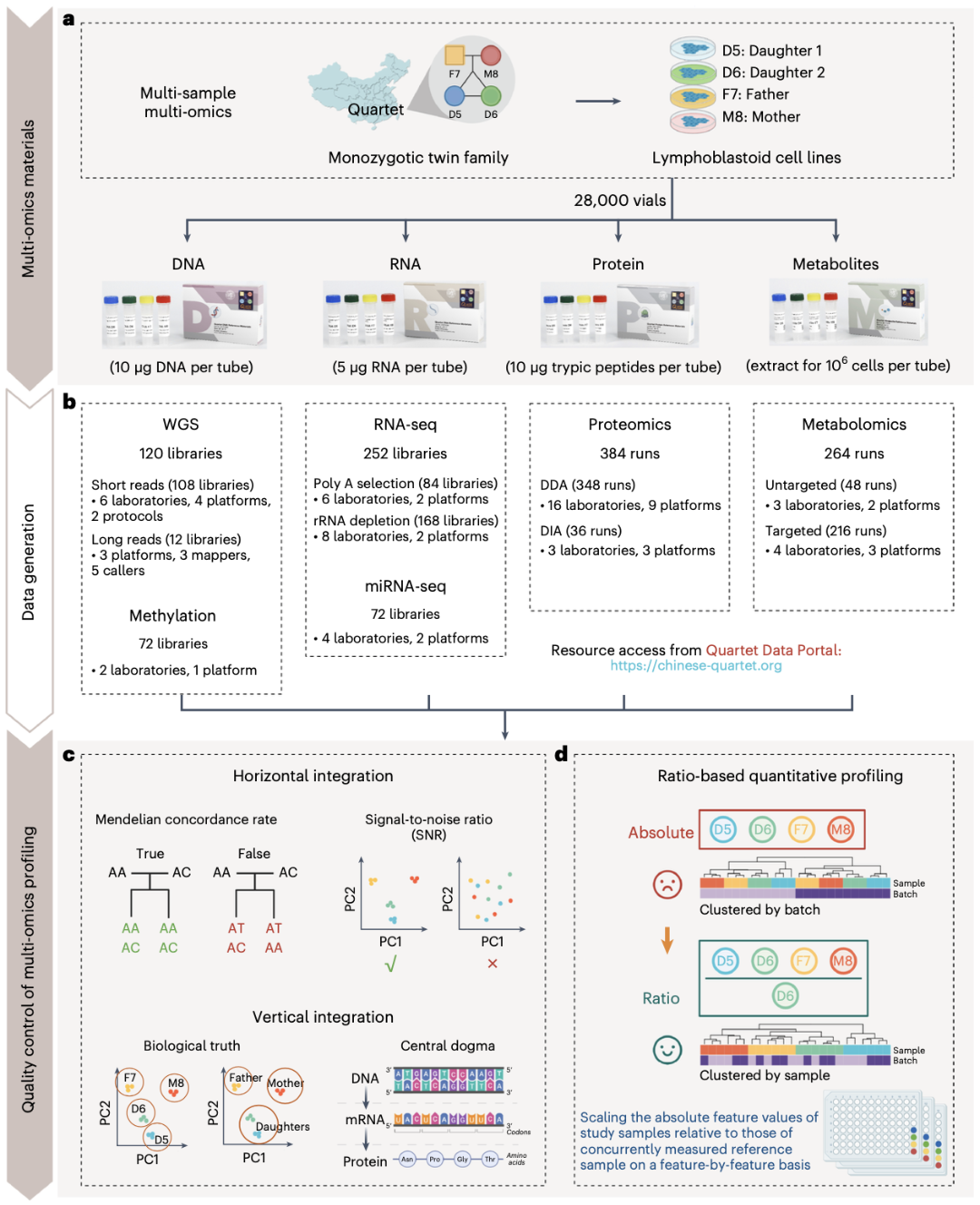
图4:Quartet多组学项目概览
RNA标准物质有效提高临床应用中检测差异表达的能力
RNA测序(RNA-seq)是转录组差异分析的常用技术,广泛应用于生物医学研究中,以发现临床诊断、预后和治疗的生物标志物。随着基于转录组的生物标志物发现成果不断涌现,RNA-seq技术将逐步成为临床常规检测项目,例如通过检测差异基因表达辅助临床治疗决策。这对RNA-seq的检测结果提出更高的可靠性要求,以提高疾病亚型间较小的差异表达的能力,提高临床差异表达的检测准确性。
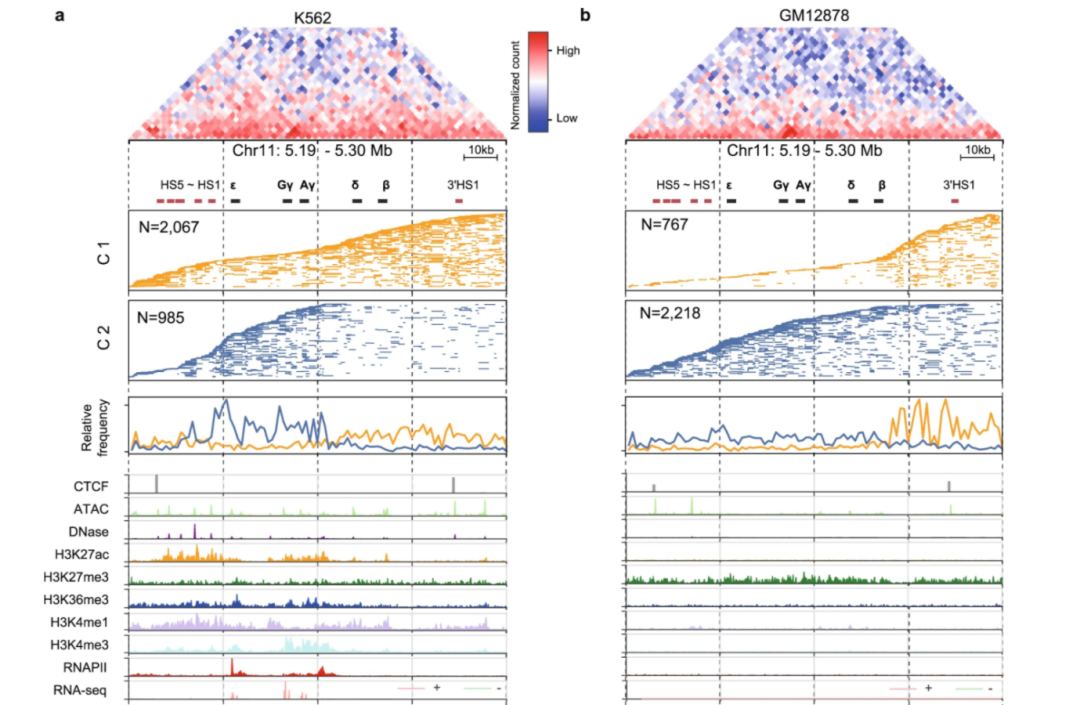
在本次发表的论文“中华家系1号”RNA标准物质与基于比值的分析方法提高了转录组数据的质量”中,研究团队指出,RNA标准物质是评估RNA-seq数据可靠性的宝贵工具,可在实验室批次内有效性和跨批次可重复性两方面对其可靠性进行客观评估。批次内有效性是在相同批次或实验室内的分析结果达到技术所能够达到的最佳水平,而跨批次可重复性是不同平台、实验室或批次间分析结果可重复,并且不受批次效应影响,跨批次数据整合后的结果与单批次结果可重复。“中华家系1号”RNA标准物质,具有微小的样本间差异、高度稳定性、长期可用性和易于生产性等特性,可用于临床应用场景下的能力测试和方法验证。
研究团队整合了不同文库构建策略、不同实验室、时间生成的21个批次RNA-seq数据集,在全转录组水平构建了基于比值的标准数据集,提供了跨平台和跨实验室数据评估的“基准”。此外,研究团队发现“中华家系1号”样本之间微小的内在生物学差异可为跨批次的RNA-seq数据整合提供高灵敏度的可靠性评估。该研究表明“中华家系1号”RNA标准物质和标准数据集,可作为评估和提高临床和生物学领域中转录组数据质量的独特资源。
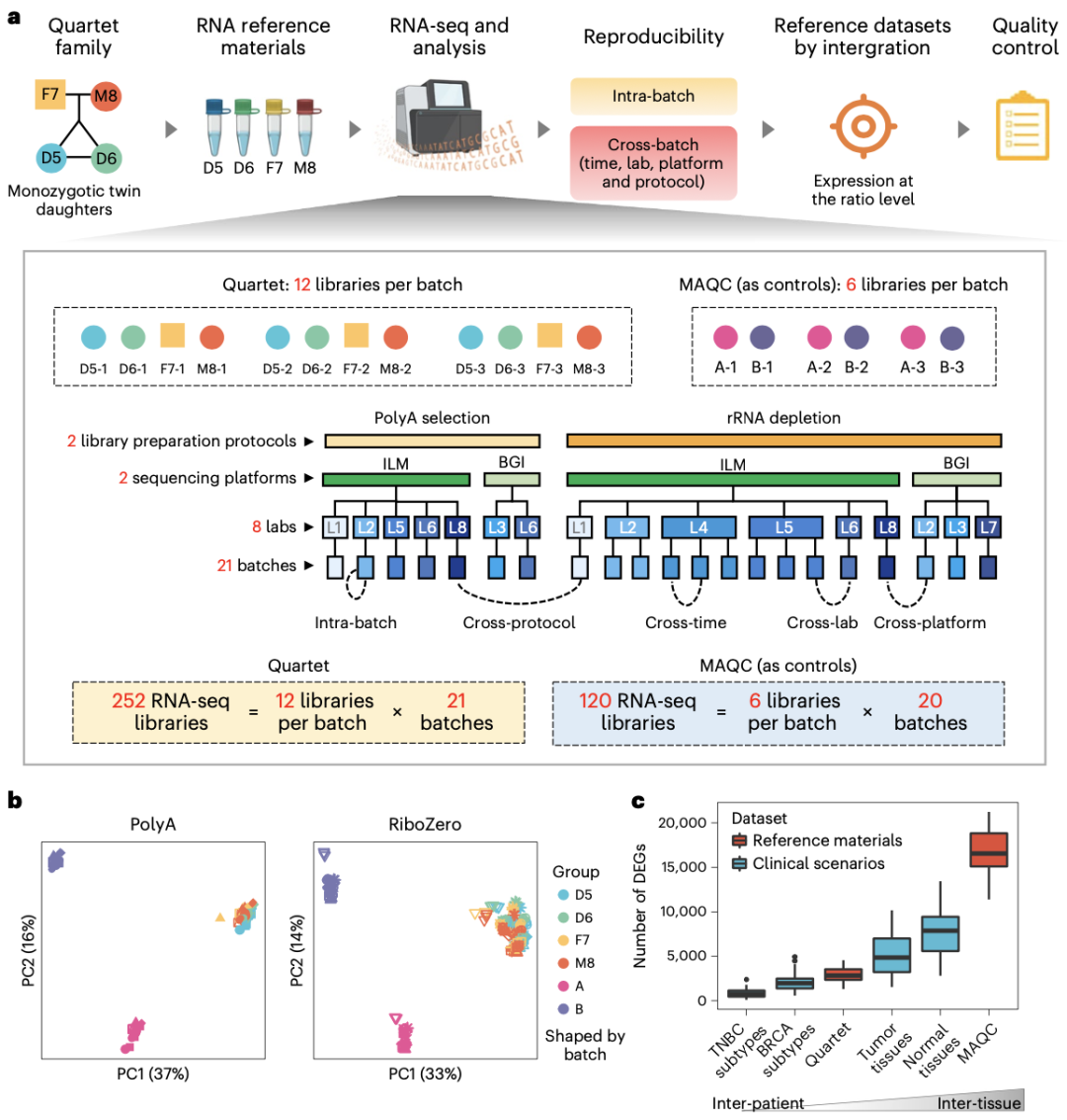
图5:Quartet RNA标准物质项目:以MQAC Sample A/B样本为参照,证明了”中华家系1号”样本间具有微小的固有生物学差异
相对定量可有效提高跨批次、跨实验室、跨平台数据的可重复性
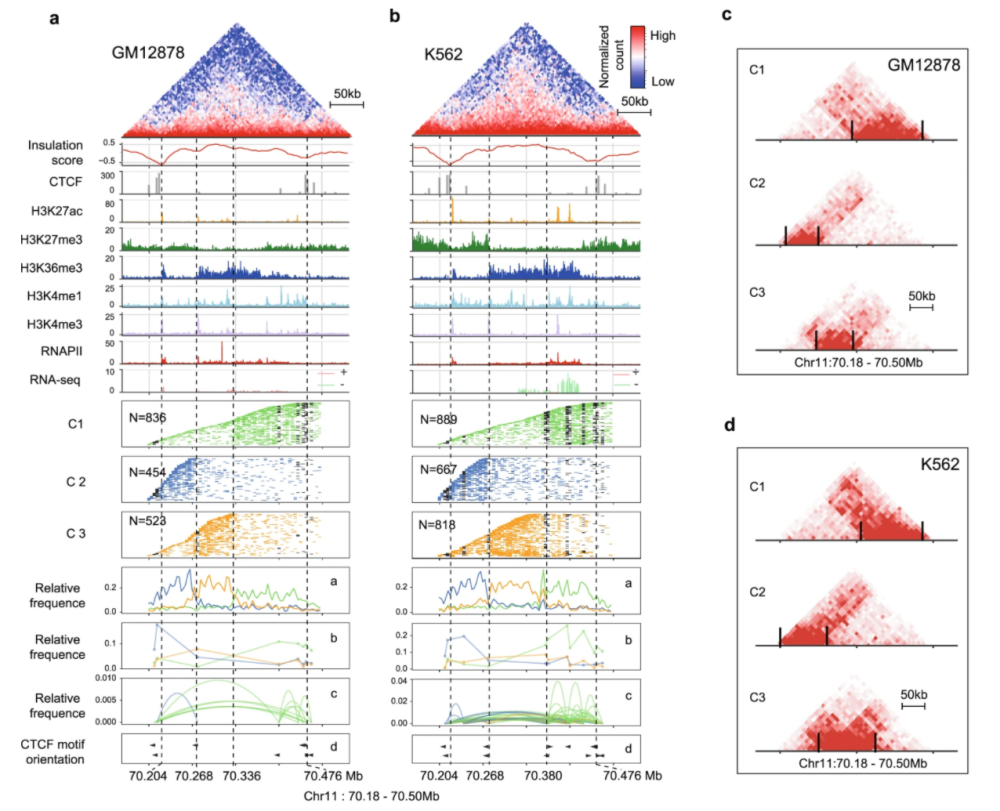
在此次发表的2篇最新论文中,中国团队取得一个重要理论性突破,那就是发现和揭示了绝对特征定量是多组学测量和数据整合不可重复性的根源,证实了基于标准物质的比值相对定量可以有效提升数据整合的质量。这对推动从绝对定量向相对定量的范式转变,实现大规模多组学数据的有效整合利用,具有重要的里程碑意义。
不同批次和平台的绝对定量多组学数据存在较大技术变异,主要受批次效应影响,无法有效反映样本间的真实生物学差异,导致数据整合效果较差。为解决此问题,研究提出一种基于比值的相对定量策略:在每个批次内使用相同标准物质作为参照,将样本的特征表达水平转换为相对于标准物质在该特征上表达的比值。
这种相对定量方法可以显著减少技术变异,提高不同批次数据之间的可比性。基于这种相对定量数据,批次效应大幅减弱,样本分类和特征关联的识别准确性显著提高,能更好反映样本间的生物学差异。特别地,主流算法难以有效校正不平衡设计下的批次效应,而相对定量方法可以有效解决。
Multi-omics data integration using ratio-based quantitative profiling with Quartet reference materials

原文链接:https://www.nature.com/articles/s41587-023-01934-1
复旦大学石乐明教授、中国计量科学研究院方向研究员、国家卫生健康委临床检验中心李金明研究员、复旦大学丁琛教授、郑媛婷副教授为本论文共同通讯作者。复旦大学郑媛婷副教授、刘雅晴、杨竞成博士、中国计量科学研究院董莲华研究员、国家卫生健康委临床检验中心张瑞研究员,以及复旦大学田莎博士为本论文共同第一作者。
Quartet RNA reference materials and ratio-based profiling for assessing and improving the quality of transcriptomic data

原文链接:https://www.nature.com/articles/s41587-023-01867-9
复旦大学郑媛婷副教授、石乐明教授、国家卫生健康委临床检验中心张瑞研究员、复旦大学钱峰副研究员和美国FDA Joshua Xu博士为本论文共同通讯作者,复旦大学郁颖青年副研究员、侯湾湾博士、刘雅晴、王海燕博士,以及中国计量科学研究院董莲华研究员为本论文共同第一作者。
相关研究得到科技部战略性国际科技创新合作重点专项“人类表型组学数据的质量控制与标准化研究”和上海市市级科技重大专项“国际人类表型组计划”资助。研究所涉及的样本和国际合作均已获得国家人类遗传资源管理部门批准,相关数据开放获取已在国家人类遗传资源管理部门备案。


















 图
图 图
图 图
图