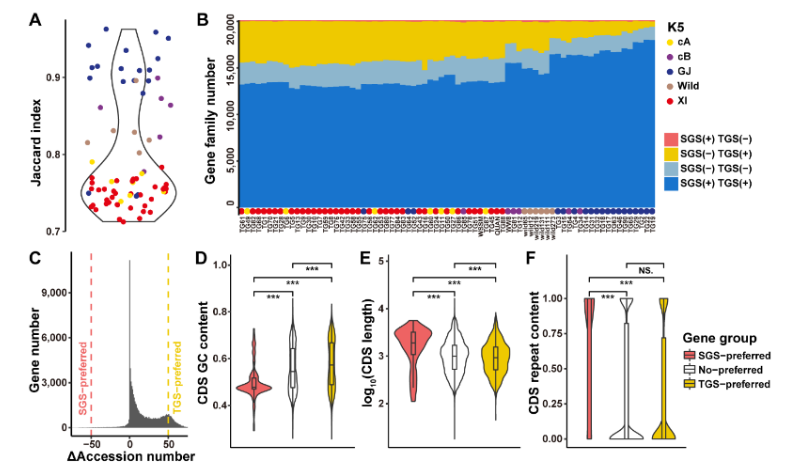
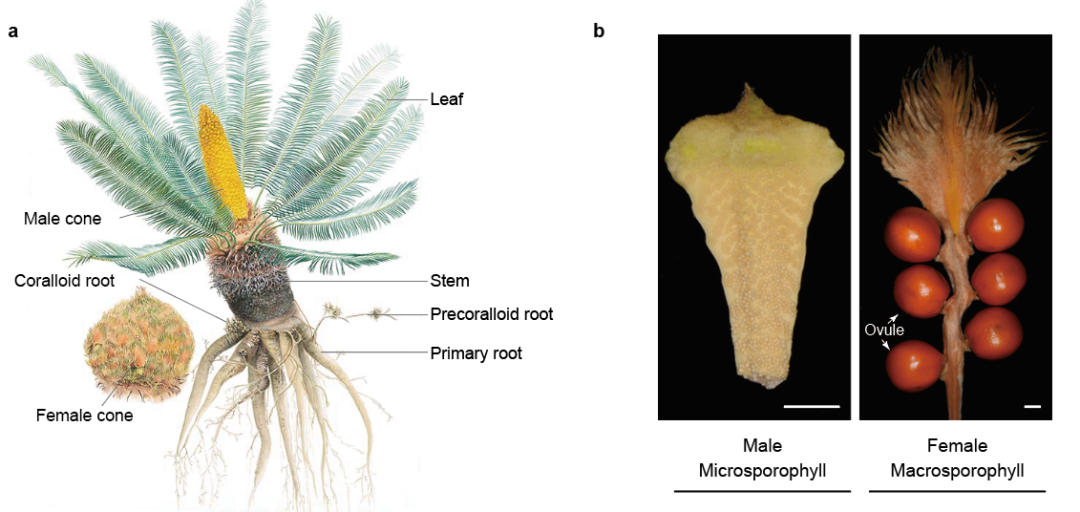
燕麦作为谷物中最好的全价营养食品,因其富含蛋白质、不饱和脂肪酸以及可溶性膳食纤维而广受消费者青睐。同时,栽培燕麦是六倍体作物,它在生物量、活力和对环境变化的适应性方面均具有多倍体植物的显著优势,在应对粮食安全挑战中可以发挥其独特作用。
北京时间2022年7月18日晚23时,国家燕麦荞麦产业技术体系首席科学家任长忠研究员领衔的以四川农业大学和吉林省白城市农业科学院为核心的燕麦研究团队在《自然-遗传学》Nature Genetics 上发表了题为 “Reference genome assemblies reveal the origin and evolution of allohexaploid oat” 的研究论文。
原文来自该项目课题组
该研究工作由四川农业大学和吉林省白城市农业科学院联合中国科学院遗传与发育生物学研究所、四川大学、西昌学院、中国农业科学院和武汉希望组生物科技有限公司合作完成,并得到了国家燕麦荞麦产业技术体系,国家自然科学基金,吉林省人才开发基金和吉林省科技发展计划等项目的资助。
四川农业大学彭远英、颜红海、邓操,吉林省白城市农业科学院郭来春、王春龙和四川大学王毓博为论文共同第一作者,吉林省白城市农业科学院任长忠研究员、中国科学院遗传与发育研究所鲁非研究员、四川大学马涛教授、四川农业大学魏育明教授和彭远英教授为论文共同通讯作者。希望组参与组装注释以及部分分析工作。
该研究发布了栽培六倍体裸燕麦及其二倍体和四倍体祖先的参考基因组,并进一步选择能代表燕麦属现存所有基因组类型的二倍体、四倍体和六倍体材料结合全基因测序、叶绿体基因组和转录组分析,深入探究六倍体燕麦的起源与亚基因组进化。

基因组组装
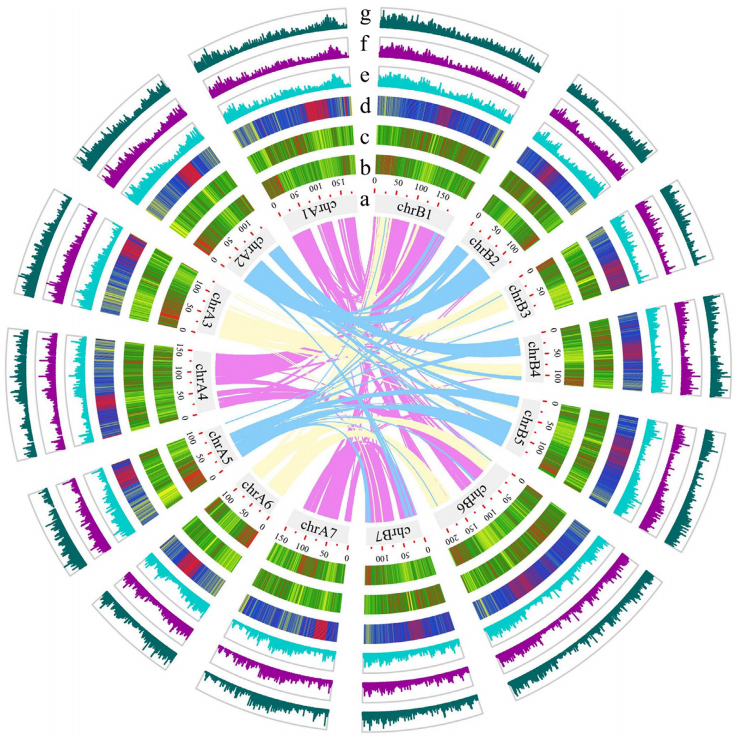
燕麦(Avena sativa L., 2n = 6x = 42, AACCDD 基因组) 作为重要的粮饲兼用型作物,由于其基因组为异源六倍体组成,基因组大(~11G)、重复序列含量高(~87%)且亚基因组间存在大量的交换,导致其基因组组装难度较大,相关研究一直滞后。研究团队利用近20年的燕麦属物种研究经验,针对六倍体燕麦亚基因组构成及其在基因组组装中可能遇到的问题,设计了利用ultra-long三代测序结合HiC、二代测序、Iso-seq和RNA-seq,并同时组装栽培燕麦最可能的四倍体和二倍体祖先的策略。项目组首先选择来自裸燕麦起源中心的传统地方品种“三分三”为材料,基于1028Gb的三代超长序列,并使用650 Gb的二代数据进行校正,组装了10.76 Gb的燕麦基因组,基于1296 Gb的Hi-C数据将99.06%的基因组序列挂载到燕麦21条染色体上(表1)。基因组组装从contig N50(75.27Mb),LAI(18.34)、BUSCO(99.44%)以及与来自六倍体燕麦一致性图谱标记的共线性等多方面进行质量评估,均显示了所组装基因组的高质量。为了准确区分亚基因组并阐明六倍体燕麦的多倍化历史,我们对其最可能的祖先物种A. longiglumis(2n=2x=14,AlAl基因组)和A. insularis(2n=4x=28,CCDD基因组)同时进行了测序和组装,分别构建了3.74 Gb和7.52 Gb的二倍体和四倍体参考基因组。基于这三个物种染色体之间的相似性,我们明确了六倍体燕麦21条染色体的A、C和D亚基因组归属,完成了六倍体的染色体组装、挂载和注释,获得了燕麦染色体级别的高质量参考基因组(图1)。
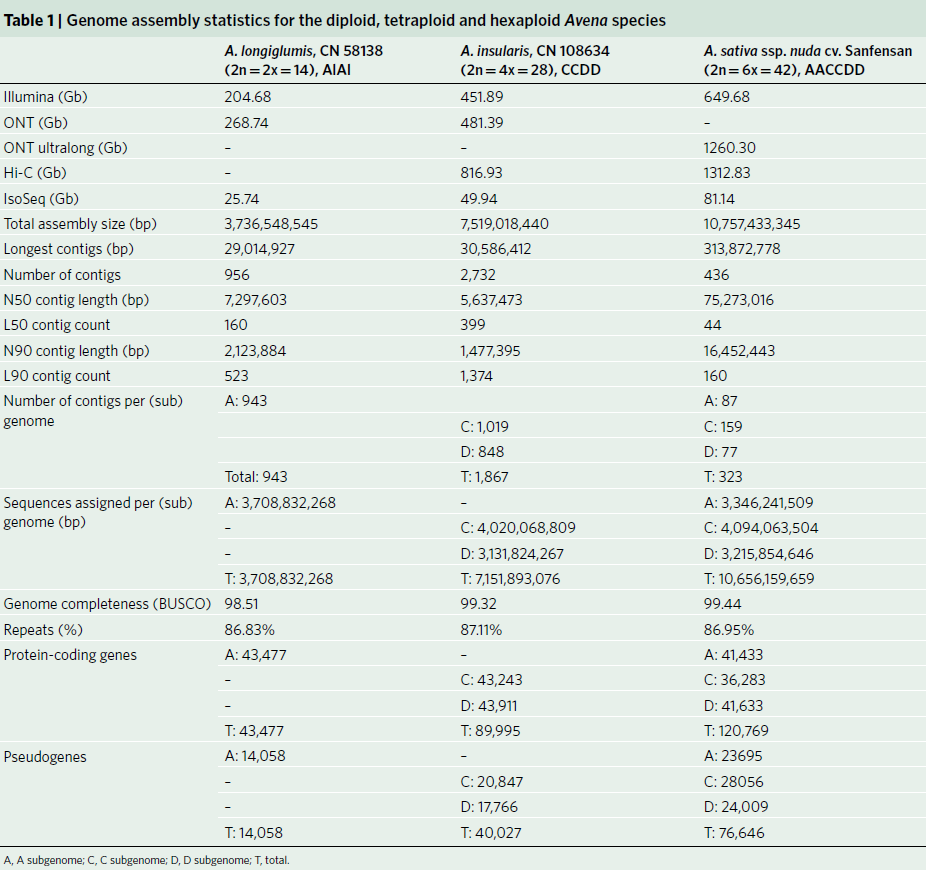

图1 AlAl基因组二倍体、CCDD基因组四倍体和AACCDD六倍体栽培燕麦的基因组。a, A和D基因组染色体着丝粒位置;b, C基因组特异性重复序列Am1在染色体上的分布。其中,三分三的的1A、2D、3D、4D和5D染色体上富含Am1的区域是C基因组渗入;c, A基因组特异性重复序列As120a在染色体的分布;d,k-mer频率;e,串联重复(TR)密度;f, LTR密度;g,基因密度;h,染色体名称和大小。最内层显示六倍体及其祖先物种的共线性,上层的彩色表示每个六倍体染色体及其祖先染色体的共线性,灰色的下层显示六倍化后的染色体重排。
系统进化基因组分析
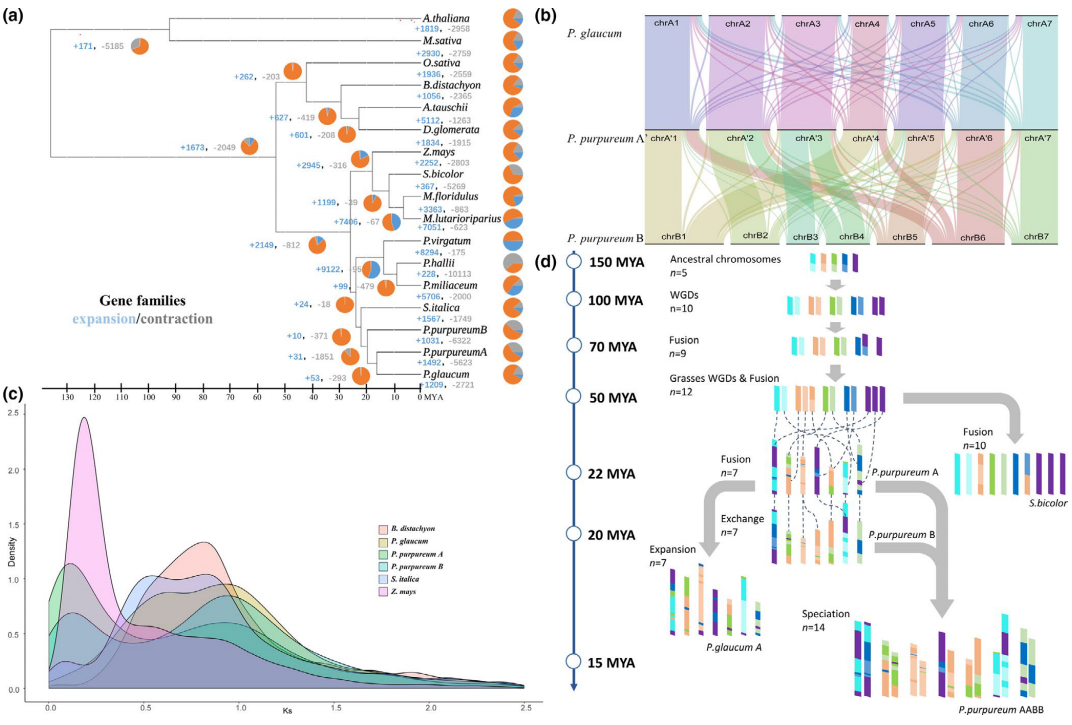
为进一步阐明燕麦在禾本科谷类作物中的进化地位,我们进行了主要禾谷类作物的系统进化基因组学分析。通过鉴定到的2237个同源单拷贝核基因对燕麦及其相关禾谷类作物的系统分析结果表明:燕麦族和小麦族之间的分化发生在稻族形成之后,且燕麦族与多花黑麦草的亲缘关系比与小麦族更近。通过与以水稻为代表的祖先核型和普通小麦的三个亚基因组进行比较,明确燕麦不同亚基因组的核型进化历史并发现在燕麦中存在大量染色体重排(图2)。
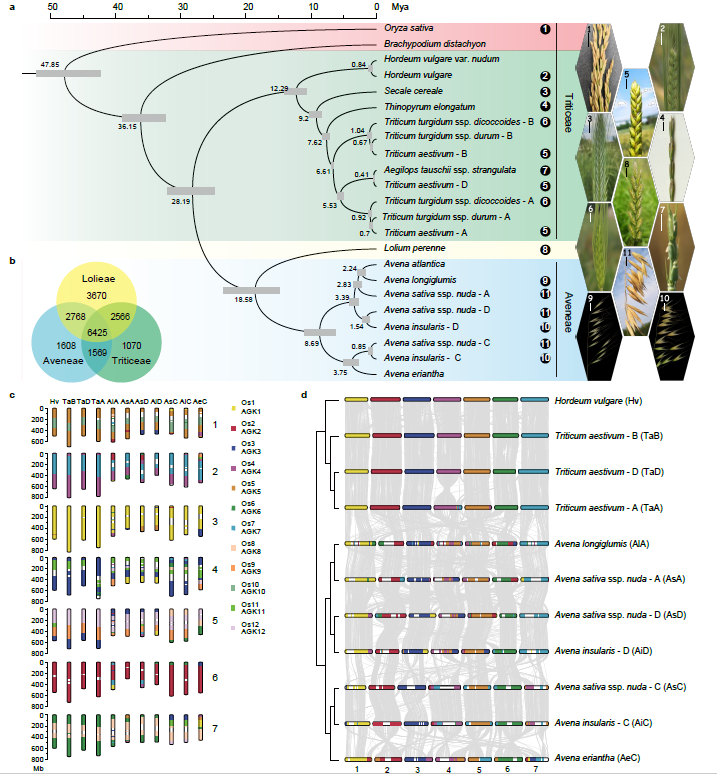
图2 禾谷物作物的系统基因组关系。a,燕麦及其相关禾谷类作物系统发育和分化时间;b,燕麦族、多花黑麦草和小麦族中共享和独特基因家族的数量韦恩图;c,燕麦和小麦可能的染色体进化核型模式。亚基因组染色体不同的颜色显示其来源于水稻不同的染色体(Os1–Os12)片段,水稻染色体可以作为祖先染色体核型(AGK1–AGK12)的代表;d,燕麦和小麦三个亚基因组之间的染色体共线性。
选择能代表燕麦属所有基因组亚型和不同倍性水平的物种进行基于全基因组重测序、转录组测序和叶绿体基因组的系统发育分析,结果一致表明Al/As基因组二倍体以及四倍体A. insularis 的C和D亚基因组分别与六倍体燕麦的A、C和D亚基因组具有最高的相似性,且通过叶绿体基因组确定D基因组是六倍体燕麦的母本供体,而C基因组二倍体作为栽培燕麦的父本基因组,通常比母系起源的基因组更容易发生染色体变异,与本研究中燕麦C基因组二倍体和多倍体物种的C亚基因组间相对较低的共线性一致。由此,我们明确了栽培燕麦的多倍化历史并提出了燕麦属物种的网状进化模式。现有的ACD基因组六倍体栽培燕麦是以Al/As基因组二倍体祖先为父本,和CD-基因组四倍体A. insularis为母本杂交加倍后形成的(图3)。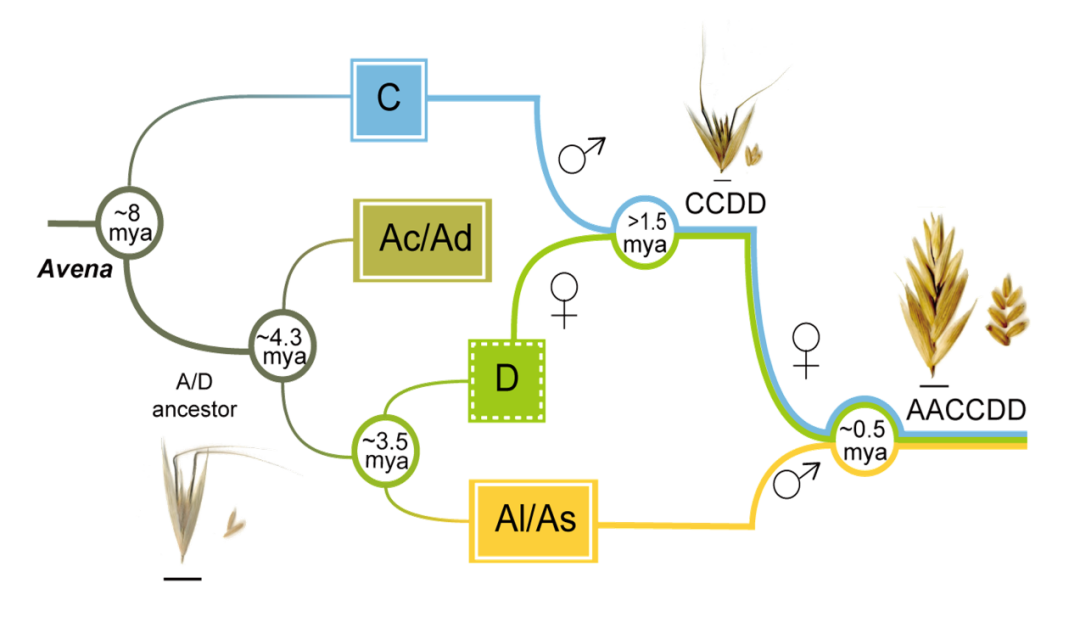
图3 六倍体燕麦的网状进化模式图
燕麦多倍化过程中的染色体结构变异
为了研究燕麦多倍化过程中发生的染色体结构变异,我们对二倍体、四倍体和六倍体物种进行了共线性分析。结果表明,在燕麦多倍化过程中发生了多次大的易位和倒位事件,我们通过荧光原位杂交证实了这些染色体结构变异(图4)。此外,在燕麦四倍体中主要是非同源染色体间的染色体易位,仅有3.91%发生在同源染色体之间,而六倍体中高达49.69%的易位是在同源染色体之间发生的,且六倍体燕麦中的同源交换有88.4%(931.94/1054.30 Mb)发生在A和D亚基因组之间,远高于A和C(11.2%,117.71/1054.30 Mb)或D和C(0.04%,4.66/1054.30 Mb),表明六倍化后的同源交换在栽培燕麦基因组结构的形成中发挥了重要作用,且在多倍体细胞核中关系更近的同源基因组更易于产生亚基因组间的重组,六倍体燕麦中A和D亚基因间相似性很高,而这两个基因组与C亚基因间的差异相对较大,因此同源交换主要发生在A和D亚基因组之间,且这两个基因组间大量频繁的染色体重组形成了类似马赛克的染色体镶嵌结构,这也是六倍体燕麦的A和D亚基因组起源问题一直存在争议的重要原因。
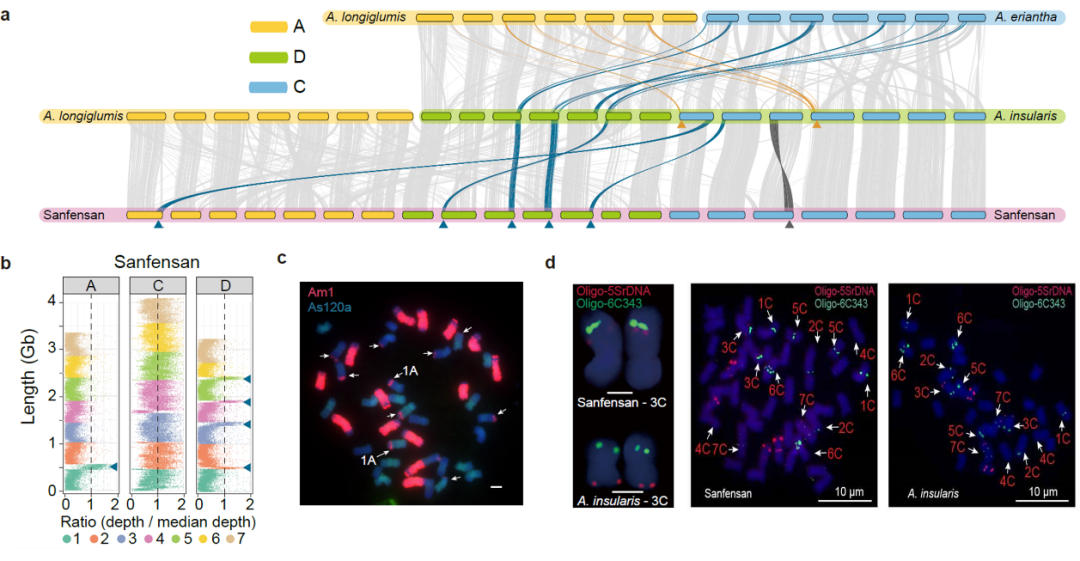
图4 燕麦属物种多倍化过程中的染色体结构变异。a,六倍体燕麦的亚基因组与可能的四倍体和二倍体祖先之间的共线性。黄色和蓝色箭头和线条分别代表观察到的来自A和C基因组的大染色体易位(>40MB)。深灰色箭头和线条表示四倍体A. insularis和“Sanfensan”之间3C染色体倒位;b,将C基因组二倍体序列比对到六倍体参考基因组显示C-A和C-D基因组间的大片段易位; c,C基因组特异性重复序列为探针的FISH证实了C-A和C-D基因组易位。来自A基因组特异性重复序列(As120a)的荧光信号以绿色显示,来自C基因组特异性重复序列(Am1)的信号以红色显示。白色箭头表示C-D和C-A基因组间易位;d, FISH验证六倍体燕麦基因组中3C染色体倒位。探针5SrDNA(红色)和6C343(绿色)分别在四倍体3C染色体的短臂和长臂上出现了明显的杂交信号,而这两个信号都同时出现在六倍体3C染色体的长臂。
六倍体燕麦中亚基因组优势
此外,多方面的证据显示六倍体燕麦在进化过程中已出现优势亚基因组(图5):首先PAV分析显示在C亚基因组中的基因丢失率较高,且C亚基因组中有更多收缩的基因家族;第二,Ka/Ks分析表明C亚基因组受到的纯化选择少于六倍体燕麦的其他两个亚基因组;第三,C亚基因组包含更多转座因子(TE),并且在基因附近显示出比A和D亚基因组更高的TE密度,而基因附近TE密度相对较高的基因往往具有较低的表达水平;第四,通过对干旱、水涝、高温、低温、盐胁迫和碱胁迫下以及燕麦不同组织的亚基因组的表达模式分析表明C亚基因组中表达的基因数量显著低于A和D亚基因组,这些结果均表明六倍体燕麦中存在亚基因组优势。
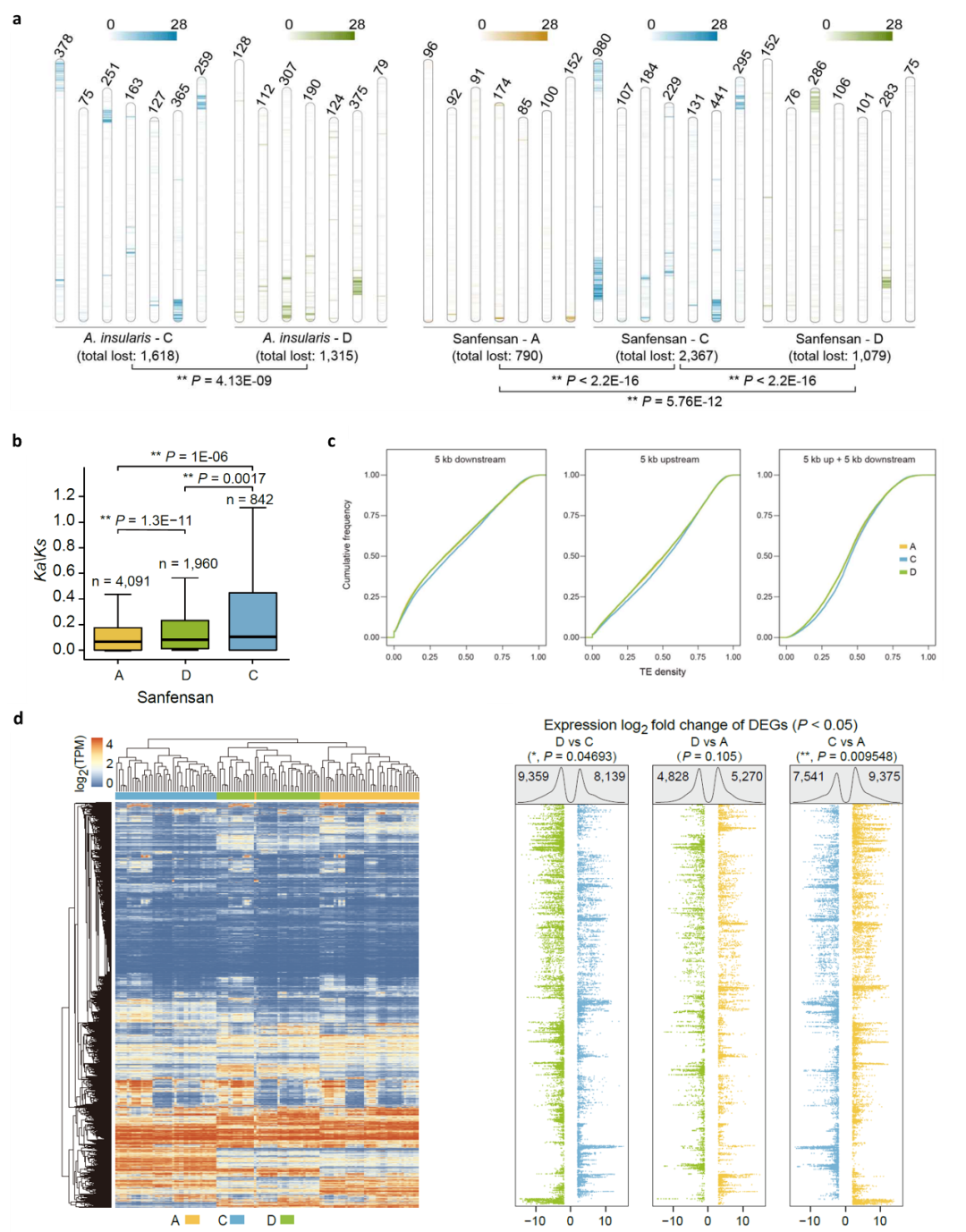
图5 燕麦属物种多倍化过程中的亚基因进化。a,四倍体和六倍体燕麦在多倍化过程中相对其二倍体亲本的同源基因丢失情况(PAV);b,六倍体燕麦三个亚基因组Ka/Ks值分布;c,六倍体燕麦三个亚基因组中基因附近TE密度的比较。相对于A和D亚基因组中的同源染色体,C亚基因组中基因附近的TE密度最高;d, 六倍体燕麦A、C和D三个亚基因组的同源基因表达差异。
燕麦抗病基因的定位和克隆
燕麦在生长中受到病害的威胁,其中最严重的是冠锈病。NBS-LRR蛋白质由一类抗性基因(R基因)编码,在植物免疫中发挥重要作用。我们在六倍体燕麦的三个亚基因组中鉴定了1269个R基因,与四倍体和二倍体不同亚基因组中鉴定的数量相比显示出收缩。大多数R基因在染色体端部成簇出现,且与燕麦已知的冠锈病基因QTL区段共定位,表明本研究构建的燕麦参考基因组可以为燕麦抗病基因的定位和克隆提供有效的参考(图6)。
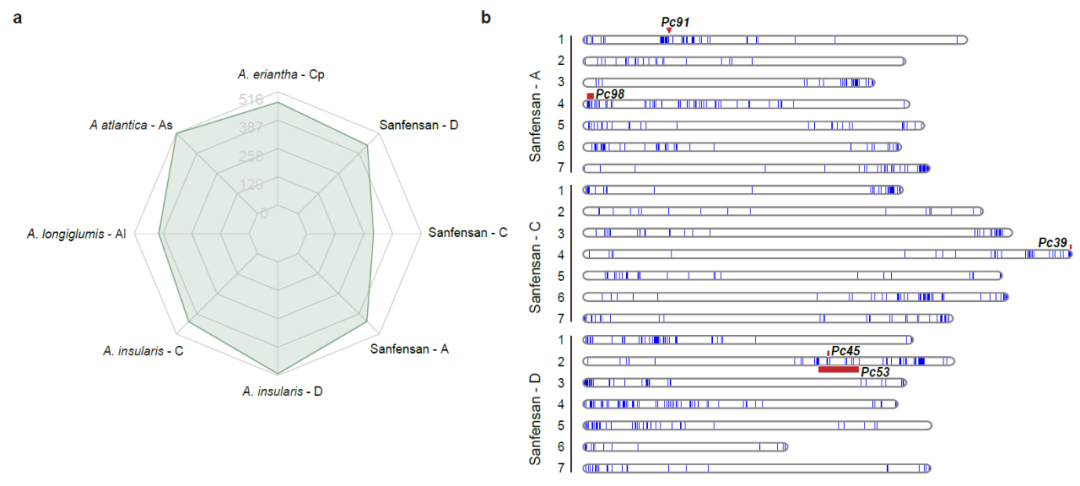
图6 | R基因在燕麦基因组中鉴定及其在染色体上的分布。a, 比较六倍体燕麦及其假定祖先基因组中R基因数量;b,冠锈病抗性QTL位点和R基因在六倍基因组染色体上的分布。
燕麦的皮裸性状
栽培燕麦根据其籽粒外壳包被情况,分为皮燕麦和裸燕麦两类。燕麦籽粒的皮裸性状是重要的驯化性状之一(图7a)。本研究通过对659份不同来源地的栽培燕麦的49702个SNP进行了全基因组关联分析,在染色体4D末端检测到显著关联区域,与之前报道的控制皮裸性状的N1基因位点区域一致。候选基因预测筛选到一个注释为受体样激酶(RLK)的基因,该基因在拟南芥中的同源基因AtVRLK1参与次生细胞壁加厚,在水稻的同源基因mis2的突变体中显示开壳小穗。比较该基因在皮、裸燕麦中的编码序列差异,发现在第一个外显子中的SNP会引起氨基酸变化。根据该SNP位点开发竞争等位基因特异性PCR(KASP)标记验证了其与皮、裸性状的相关性;通过比较10个皮燕麦和12个裸燕麦之间的转录组数据,我们发现该基因在裸燕麦中高表达;同时,在燕麦不同发育阶段的穗部表达模式也表明该基因在裸燕麦穗部发育过程中的表达量远高于皮燕麦。这些结果表明A.satnudsfs4d01g00045是控制燕麦的皮裸性状可能的候选基因(图7)。
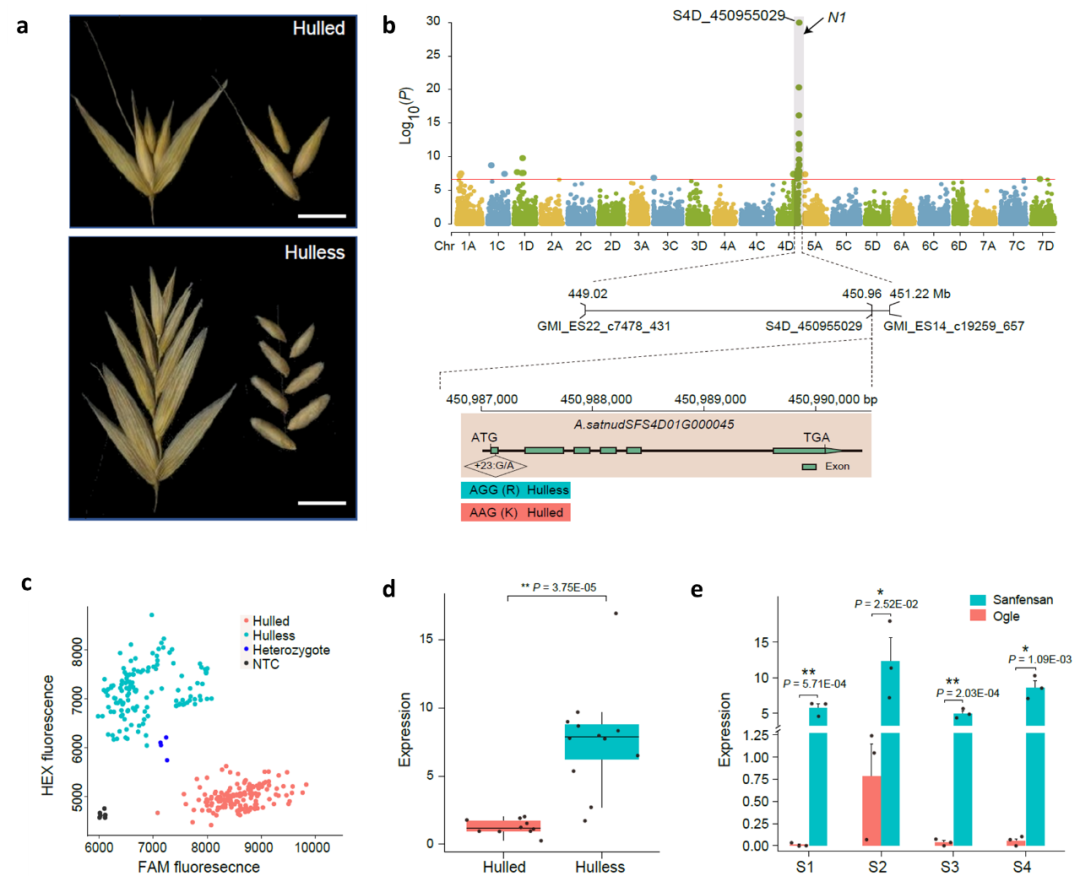
图7 燕麦裸粒性状的全基因组关联分析及候选基因预测。a,皮裸燕麦的小穗和籽粒;b,659个燕麦品种全基因组关联分析N1候选区域关联映射的曼哈顿图及其候选基因A.satnudsfs4d01g00045的基因结构;c,基因SNP差异的KASP标记验证A.satnudsfs4d01g00045与皮裸性状之间的关联;d,A.satnudsfs4d01g00045在10个皮燕麦和12个裸燕麦七个不同组织或不同发育时期等量混合的RNA样本中的表达水平;j, A.satnudsfs4d01g00045在裸燕麦“三分三”和皮燕麦“Ogle”穗部不同发育时期的表达水平比较。S1、S2、S3和S4分别代表孕穗期(Zadok’s 45)、抽穗期(Zadok’s 50)、开花期(Zadok’s 58)和灌浆期(Zadok’s 83)的穗。



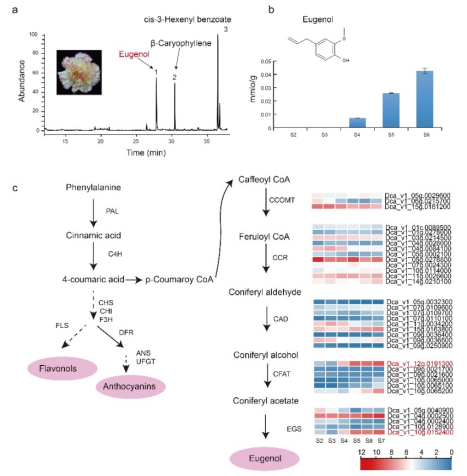
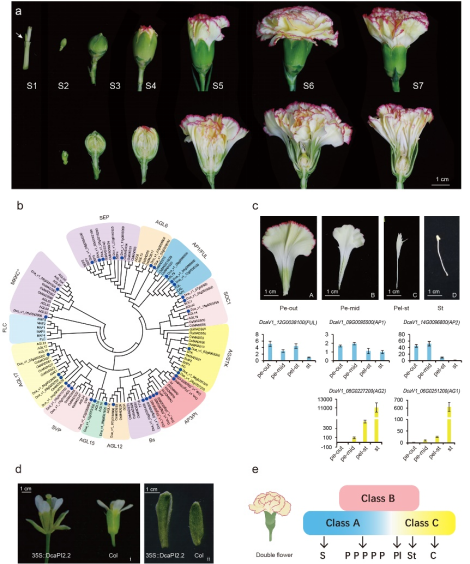
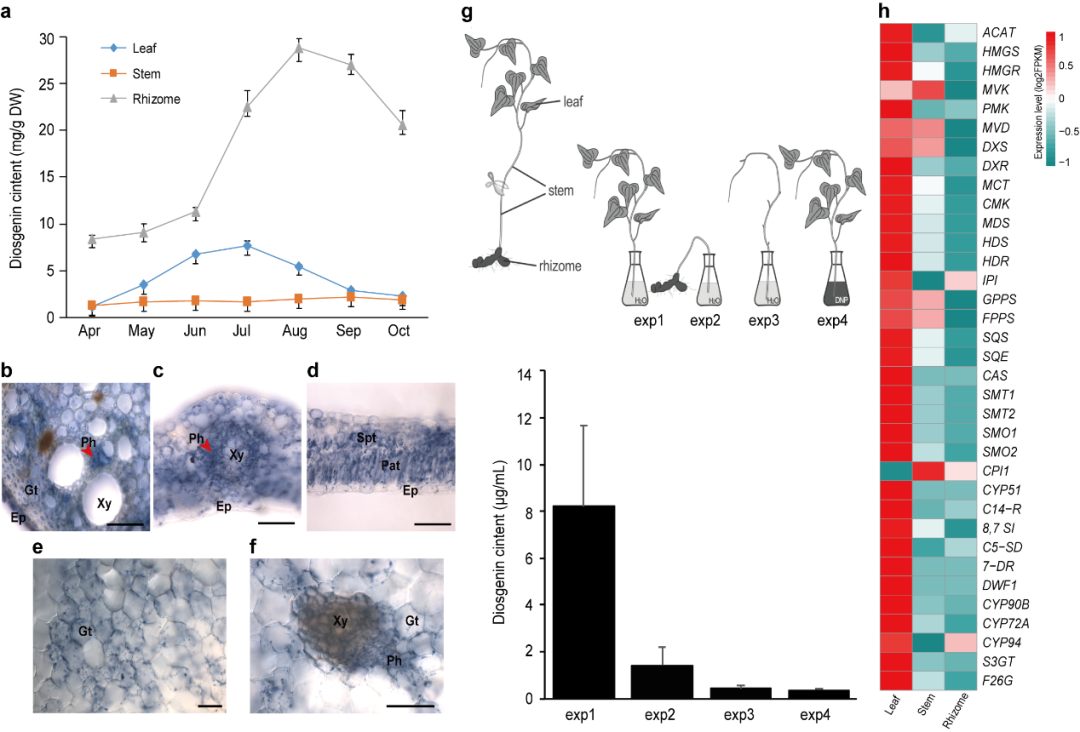 图2 盾叶薯蓣不同组织中薯蓣皂素的分布及基因表达水平比较
图2 盾叶薯蓣不同组织中薯蓣皂素的分布及基因表达水平比较





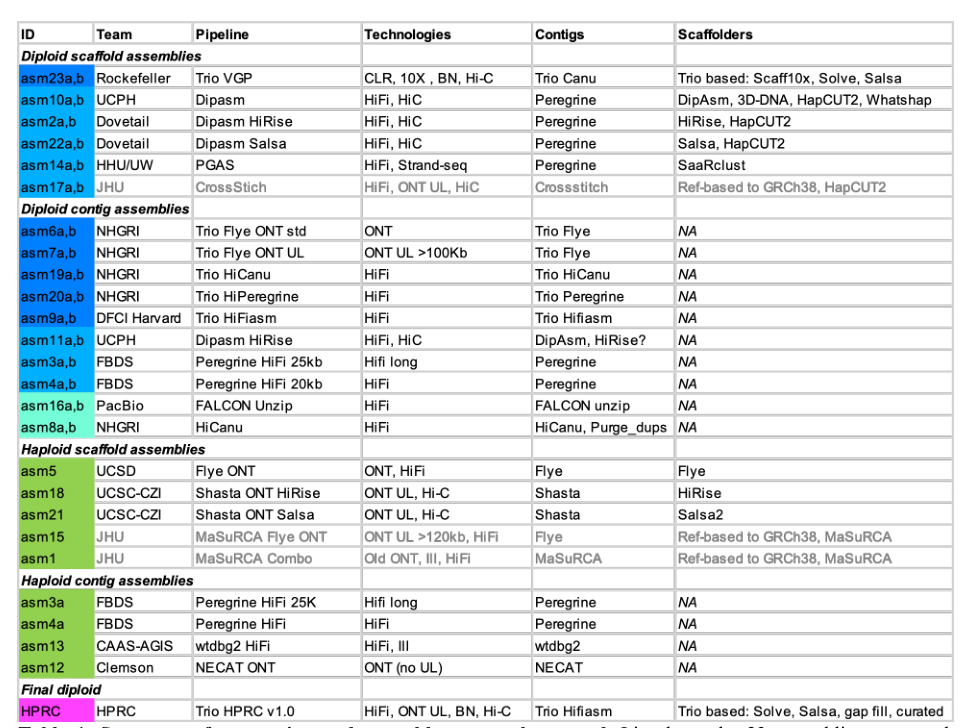
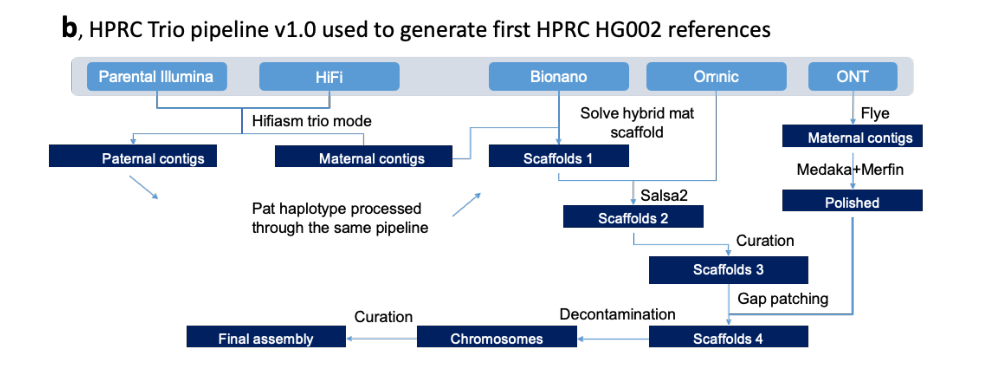 HPRC Trio pipeline v1.0组装流程图
HPRC Trio pipeline v1.0组装流程图

