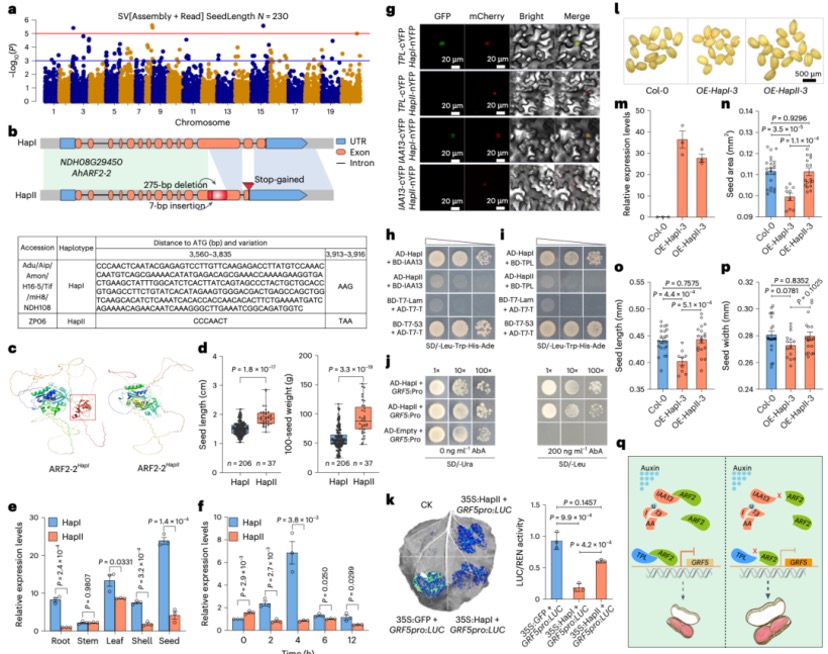
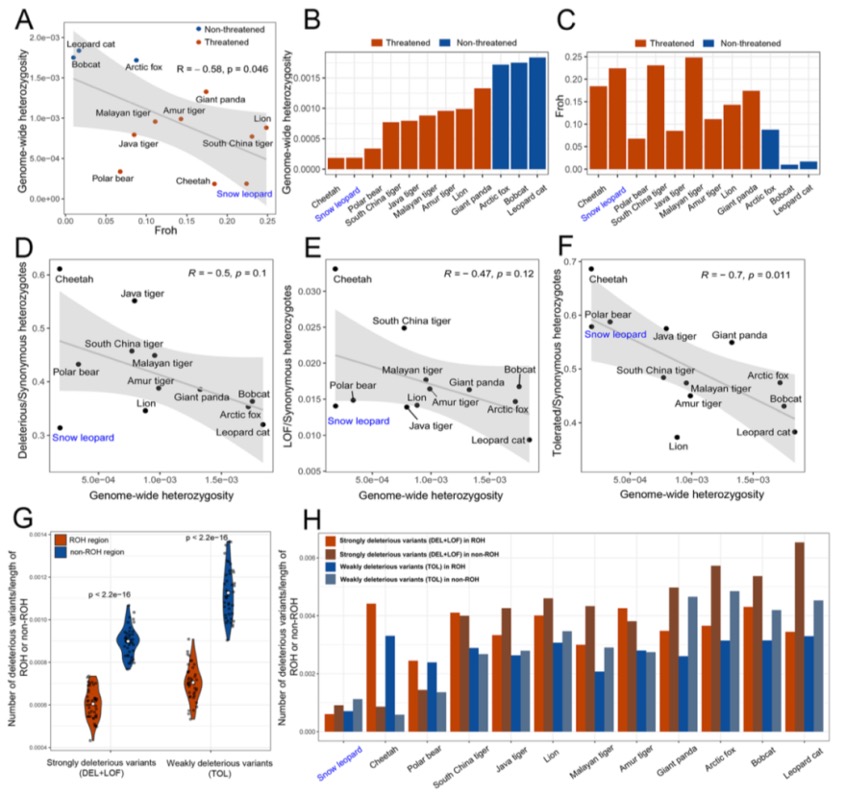
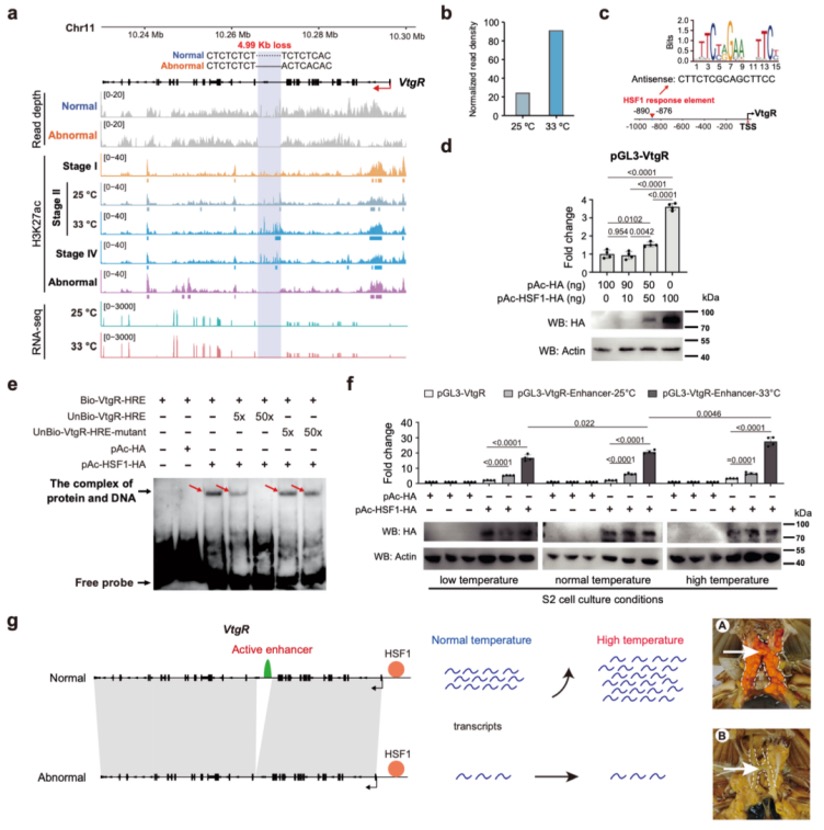
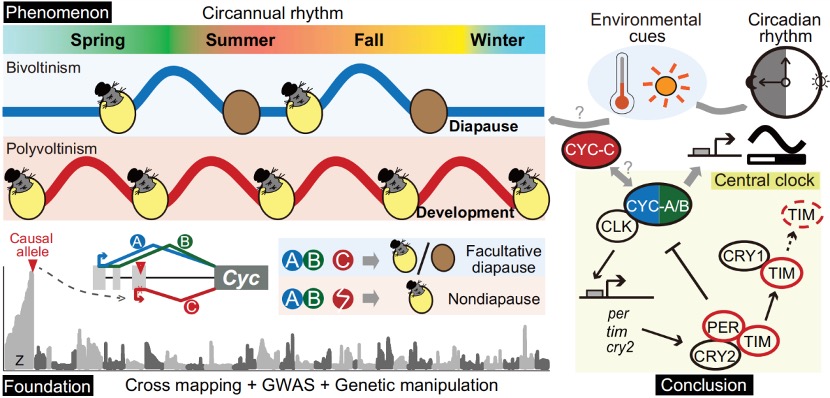
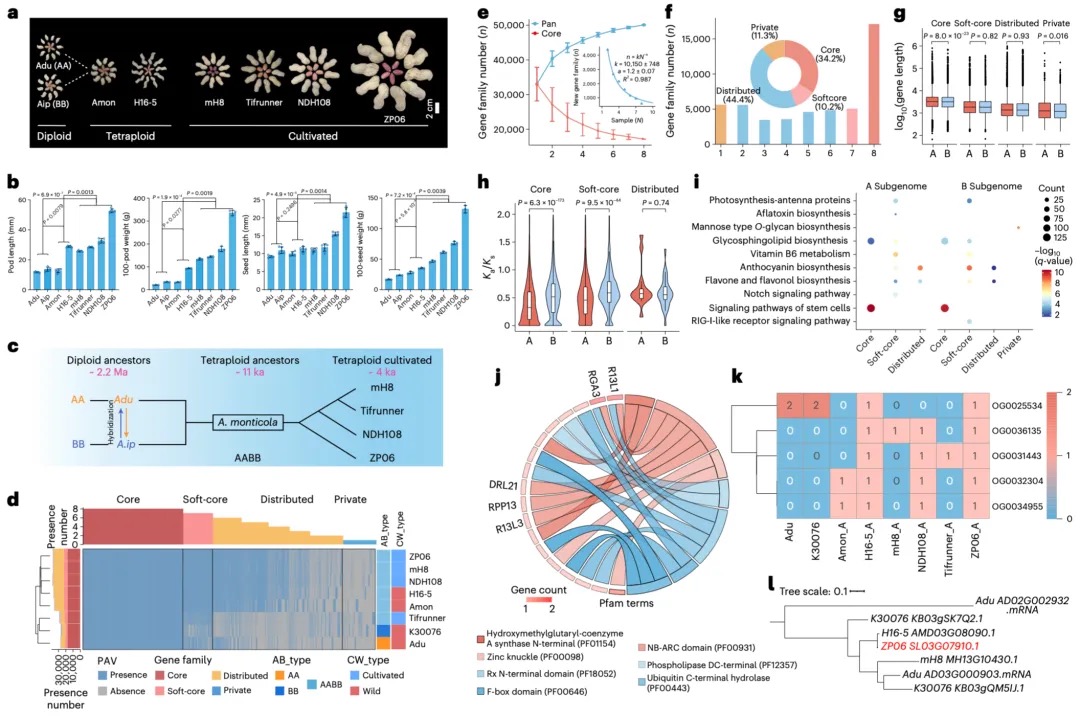
Nature Genetics署名文章 | 康乐院士团队揭秘飞蝗超大基因组背后的染色质动态调控机制
飞蝗(Locusta migratoria)具有6.9 Gb的基因组,是昆虫中大型基因组的典型代表,具有明显的密度依赖型表型可塑性和X0性别决定系统。表型可塑性是指具有相同基因型或基因组的生物体,根据环境变化改变其表型的能力,这是蝗虫的一个明显的表观生物学特征。环境的变化会促使蝗虫从散居到群居的行为转变,最终产生破坏性的蝗灾。因此,飞蝗的两型转变过程中的行为差异,是蝗灾爆发的主要原因。现有研究表明,飞蝗的群聚型行为的发生,由编码多巴胺生物合成途径中最关键酶的Henna基因决定。与果蝇等小基因组昆虫相比,飞蝗的 Henna 基因总长度增加了32倍,这主要归因于其内含子的极度扩张。因此,Henna基因是研究染色质在昆虫表型可塑性调控中作用的独特案例,特别是在扩张的内含子区域方面。
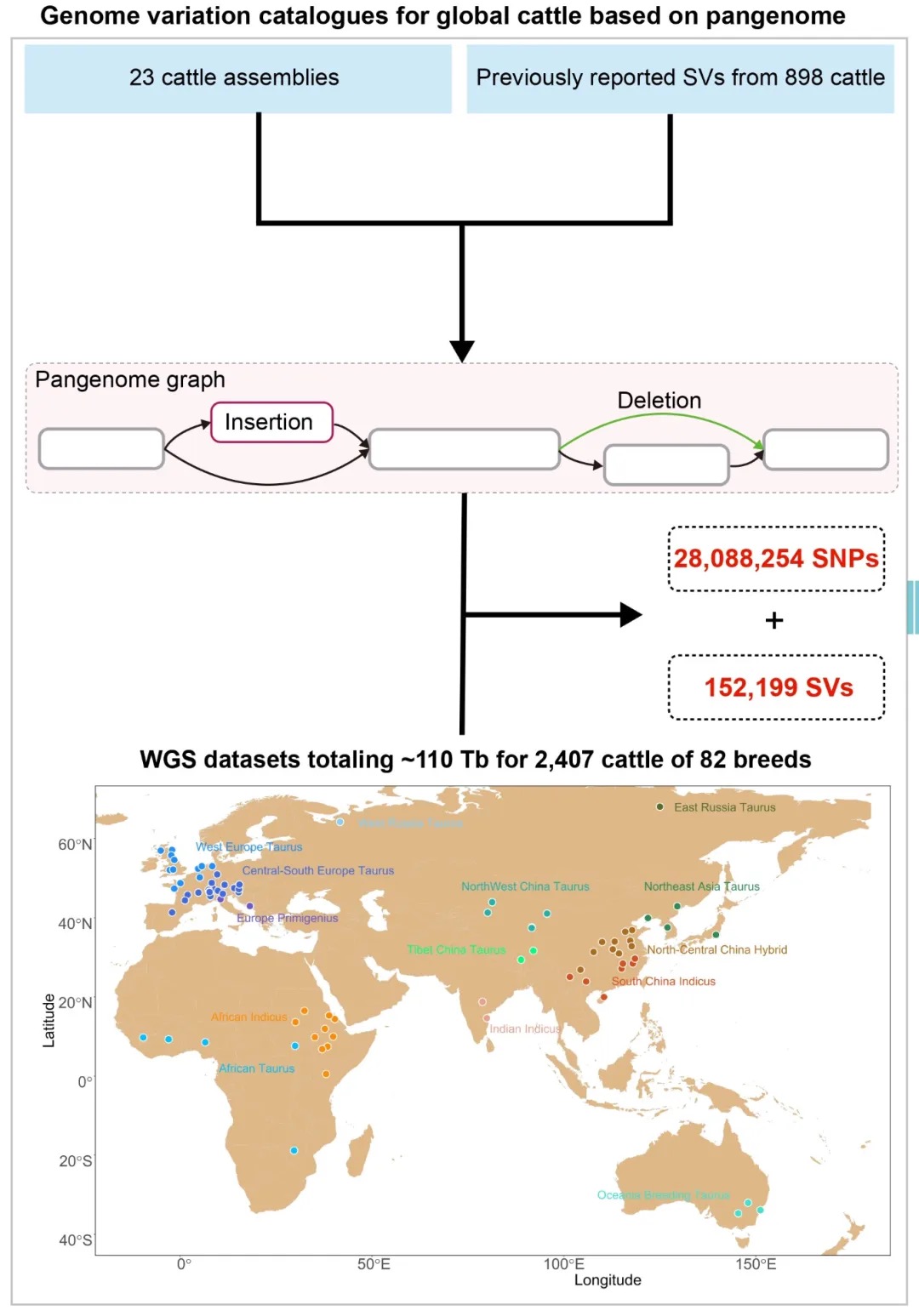
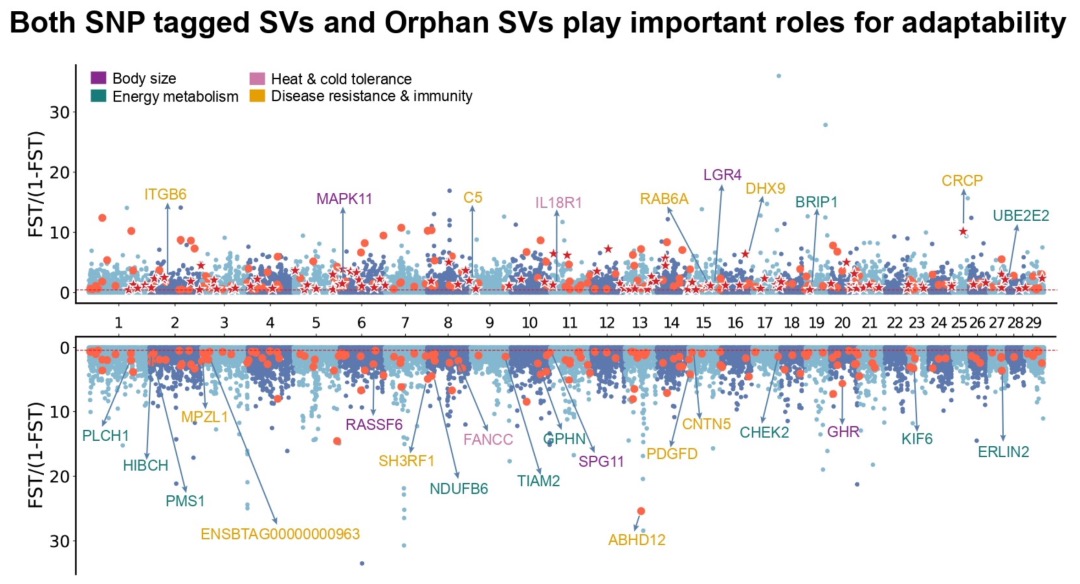
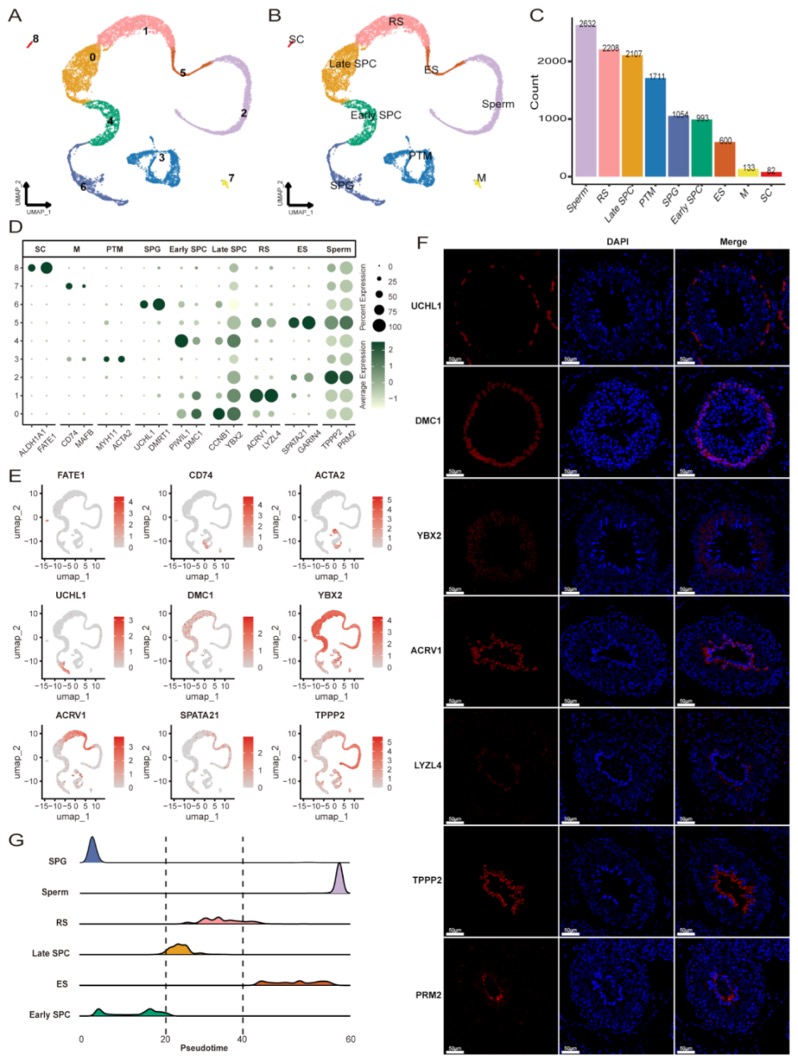
本研究采用长读长测序、光学图谱和染色体构象捕获(Hi-C)技术,对一只杂合度为1.58%的雌性飞蝗进行了基因组测序,组装版本为LMv3.1。通过Hoechst染色,成功鉴定出雌蝗中第三长的scaffold为X染色体。其在雌性中的测序覆盖度与常染色体相当,且在雄性中的覆盖度约为雌性的一半,符合X0性别决定系统的特征。LMv3.1组装版本具有很高的连续性,显著提升了转座元件(TEs)注释的完整性,并通过FISH实验验证了染色体末端结构的完整性。结合RNA表达和同源数据,预测了18,127个蛋白编码基因。与旧版本(LMv2.4)相比,新组装的全长转录本映射率更高,表明其能更准确地覆盖转录本结构。基因组质量的显著提升为深入研究蝗虫的表观基因组调控机制提供了坚实基础。
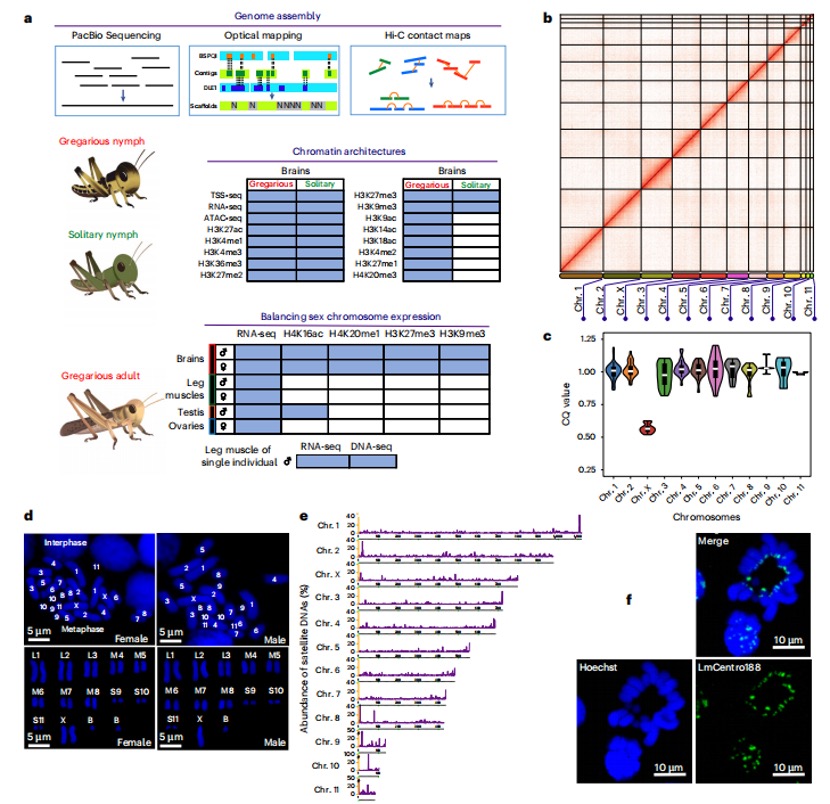
图1 飞蝗染色体级别基因组组装
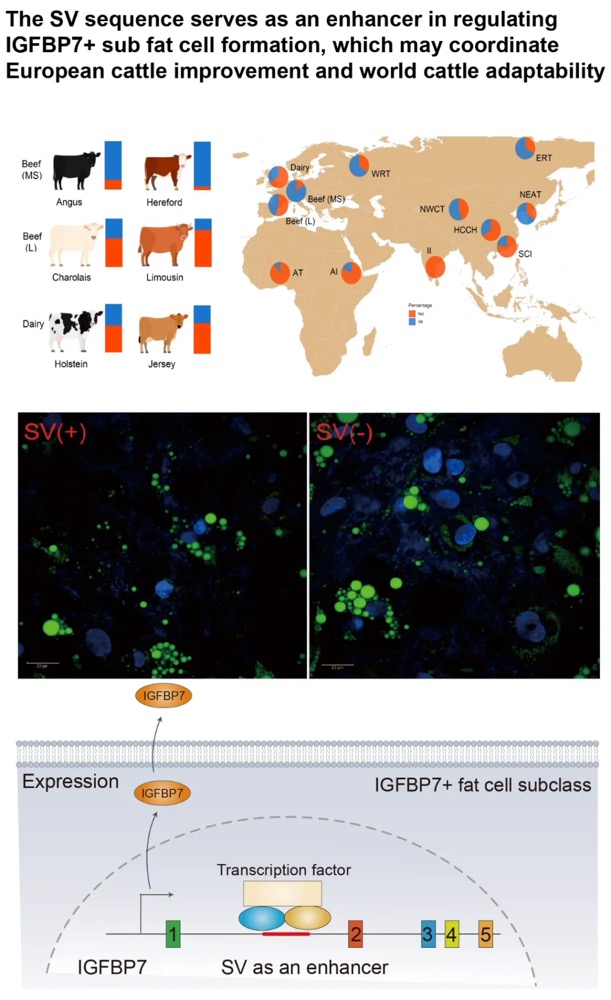
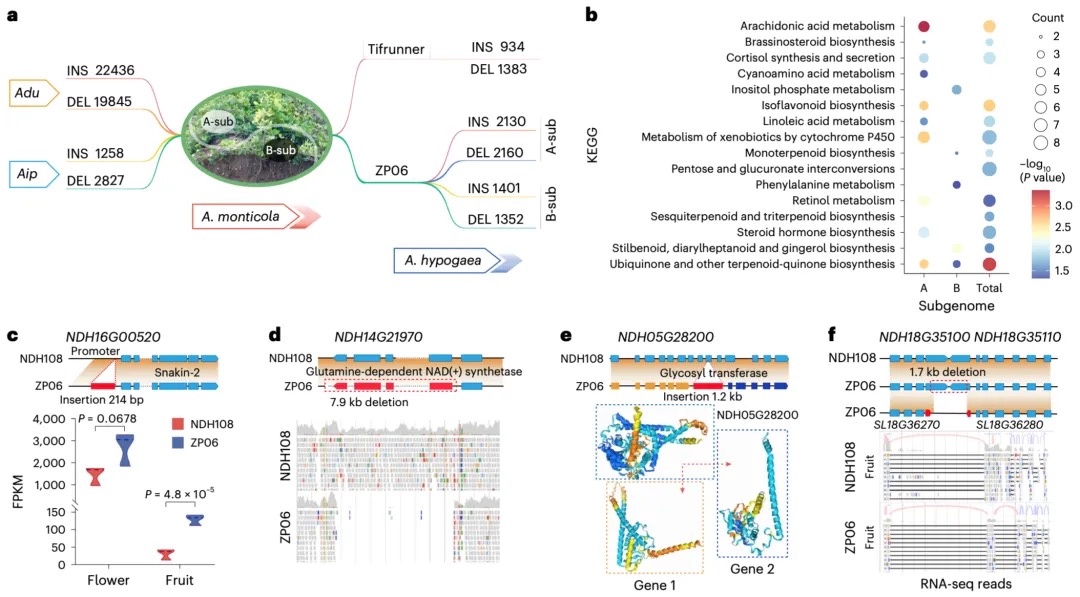
本研究通过综合运用CUT&Tag(针对13种组蛋白修饰)、ATAC-seq、TSS-seq和链特异性RNA-seq等多种高通量测序技术对脑组织染色质结构进行了多维解析。研究发现蝗虫的内含子长度显著长于其他昆虫,并且这种内含子的扩展与内含子中增强子(特别是染色质状态E5和E7)数量的增加显著相关。尽管基因长度本身的变化对基因表达水平没有显著影响,但研究发现内含子中的增强子元件对基因表达有重要提升作用,因为含有内含子增强子的长基因其表达显著高于不含此类增强子的长基因。增强子数量的增加与基因组大小的扩张共同作用,可能是在基因长度差异巨大的背景下,维持蝗虫基因表达均衡的重要机制。
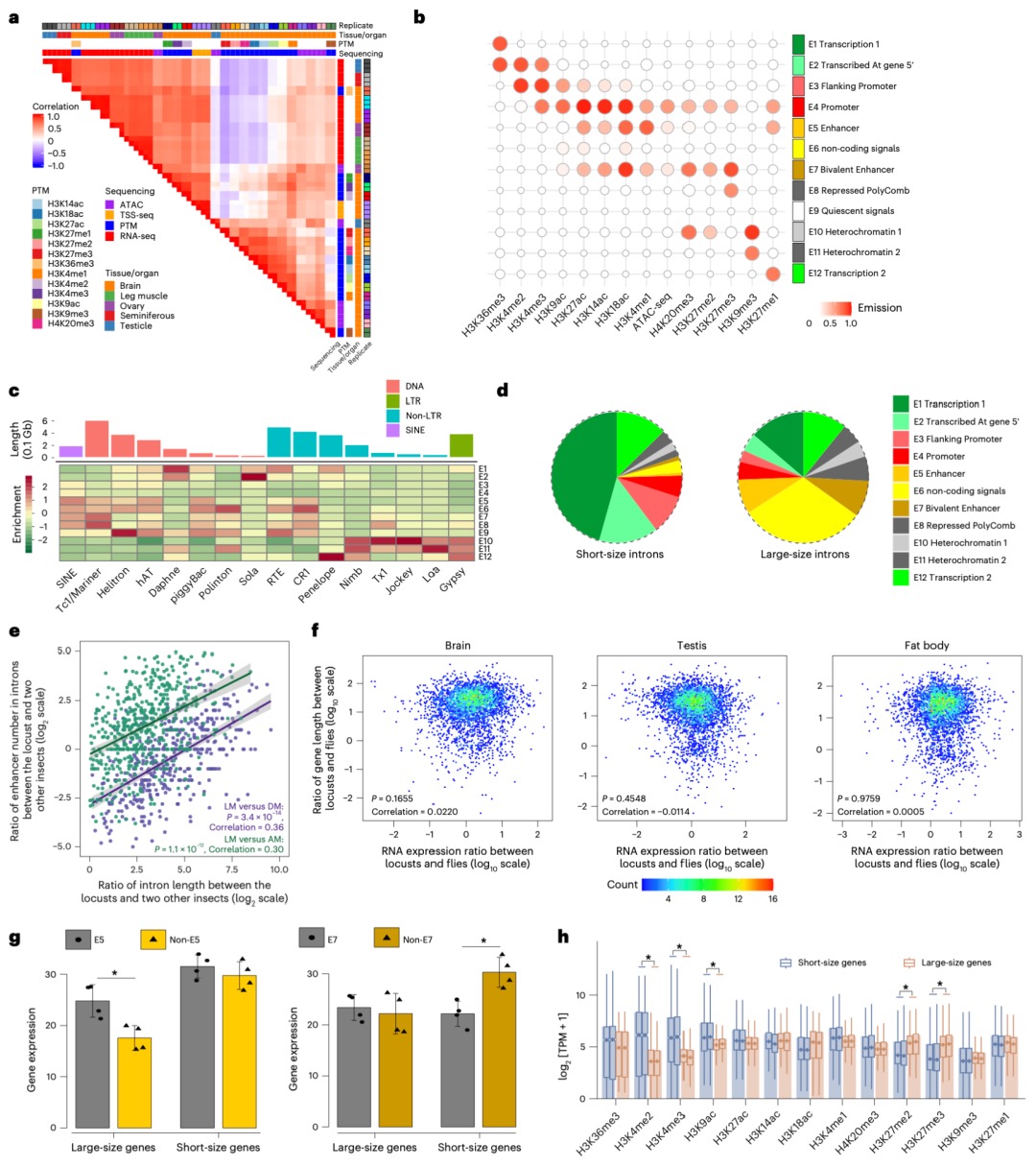
图2 长基因与短基因的增强子数量差异
本研究以蝗虫行为多型性为例,深入探讨了大基因组中表观遗传调控的机制。研究发现,群居与散居蝗虫大脑中存在显著的组蛋白修饰差异和基因表达谱变化,这些差异涉及行为调控、信号转导等重要通路。关键发现包括:H3K4me3/H3K27ac在启动子区的正相关以及H3K4me1/H3K27me3在远端区域的负相关分别调控了基因的启动和增强子活性;并进一步鉴定出Trl、Sp1和Clamp等转录调控因子可能在此过程中发挥核心作用。研究尤为重要的是,以多巴胺合成关键基因Henna为例,发现其内含子区域存在的增强子EH1(富含H3K4me1)与启动子存在三维空间互作;通过CRISPR敲除实验证实,破坏EH1会显著降低Henna表达并使群居蝗虫行为向散居型转变,这揭示了大型基因的内含子中存在的增强子通过染色质空间互作调控基因表达,是影响蝗虫行为可塑性的重要机制。
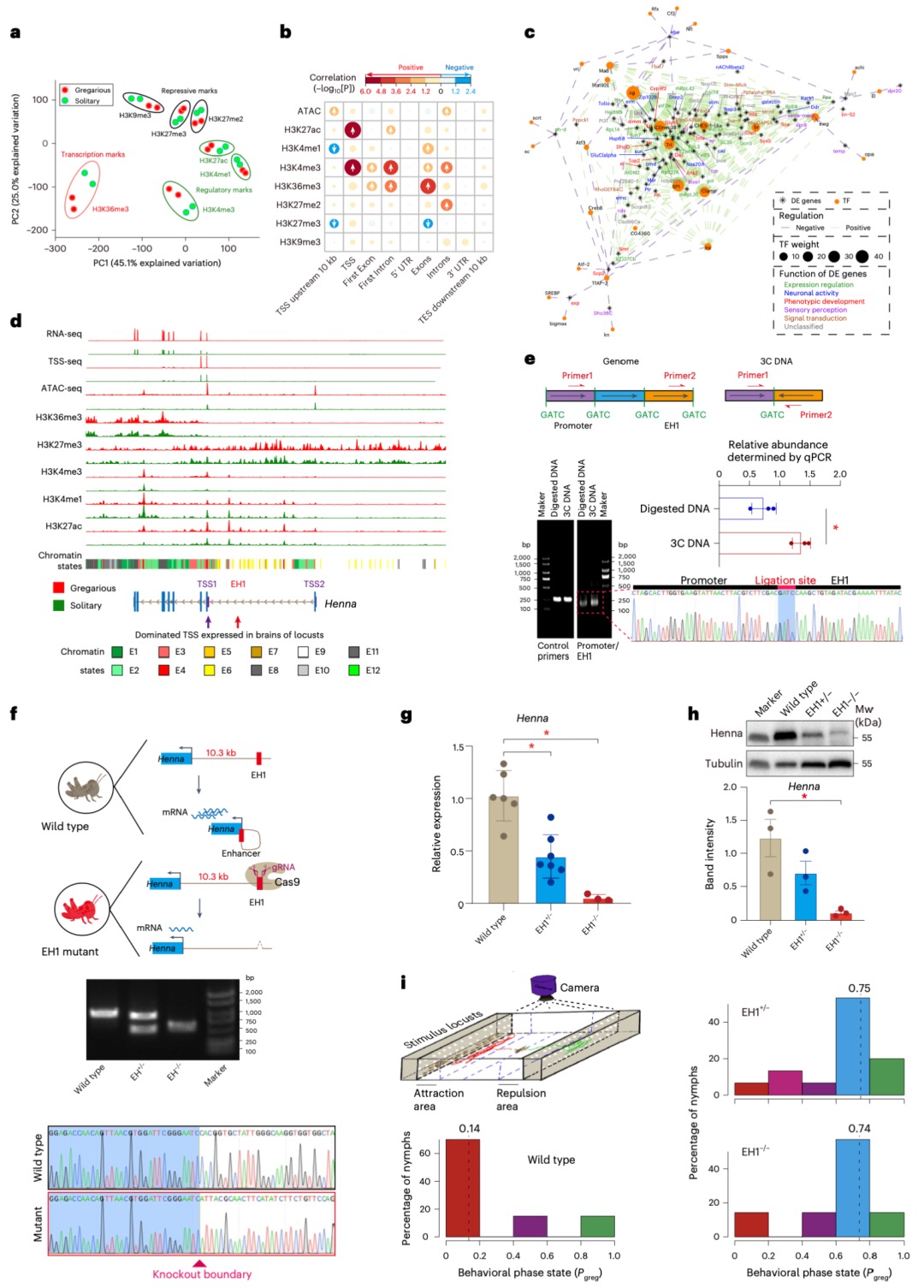
图3 蝗虫两型转变过程中染色质重塑
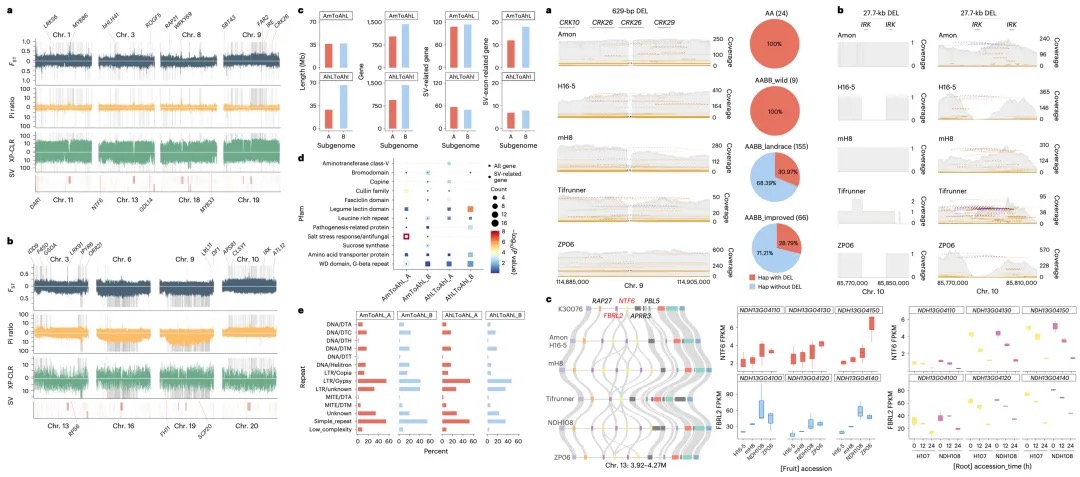
本研究发现,在蝗虫(具有X0性别决定系统)的体细胞(如大脑和腿肌)中,通过特定的表观遗传调控(如H4K16ac富集),使得单条X染色体的基因表达水平与常染色体达到相当,实现了两性间的表达平衡;然而在性腺(精巢)中,由于减数分裂性染色体失活(MSCI)的存在,X染色体表达显著降低(X/A比值~0.5),这种平衡被打破。研究进一步通过Hi-C和DNA FISH等技术揭示,失活的X染色体表现出更高的长程染色质互作频率、更压缩的高阶结构以及更远离转录活跃核中心(如核斑)的空间位置,这些独特的染色质构象特征与其基因沉默状态相关。这表明蝗虫在其大基因组背景下,采用了一种依赖于染色质高级结构组织和组蛋白修饰(如H4K16ac)的独特剂量补偿机制来调控X染色体活性,该机制在体细胞和性腺中存在显著差异。
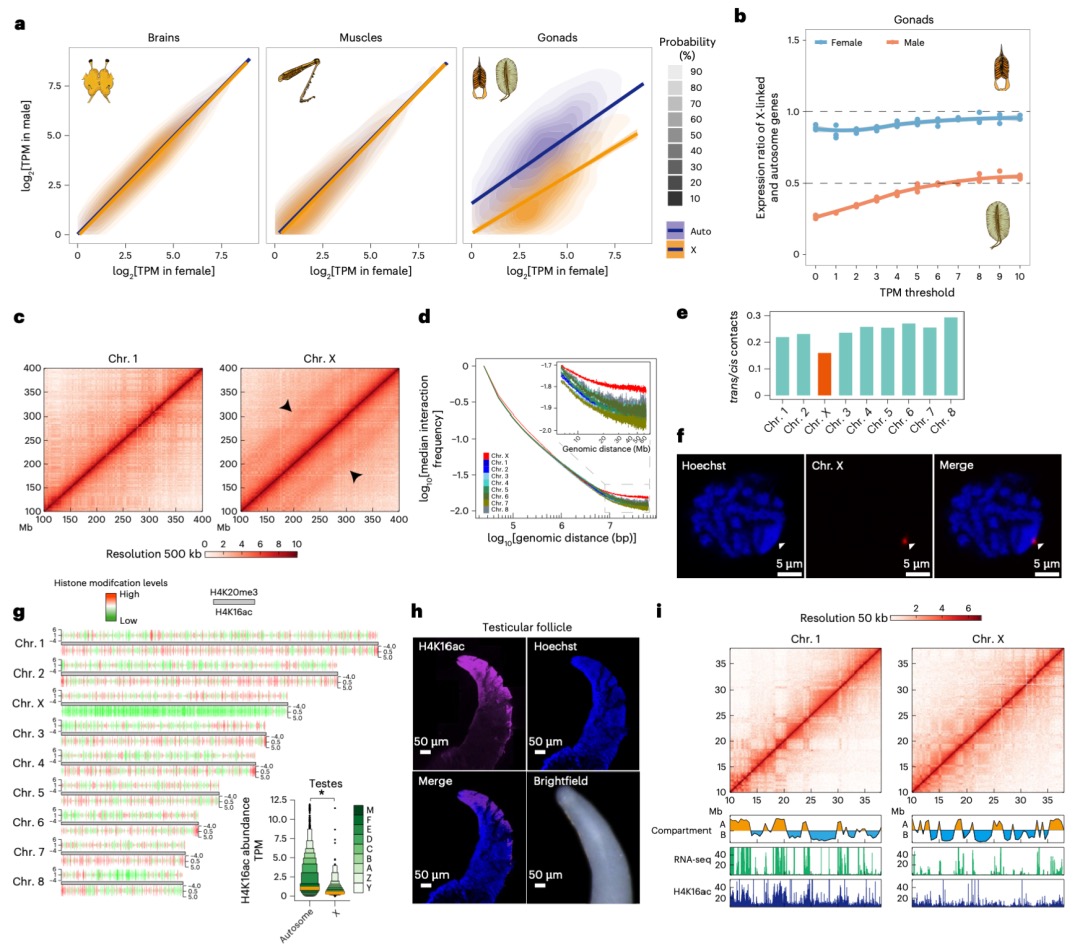
图4 减数分裂沉默中X染色体的全局互作与染色质重塑
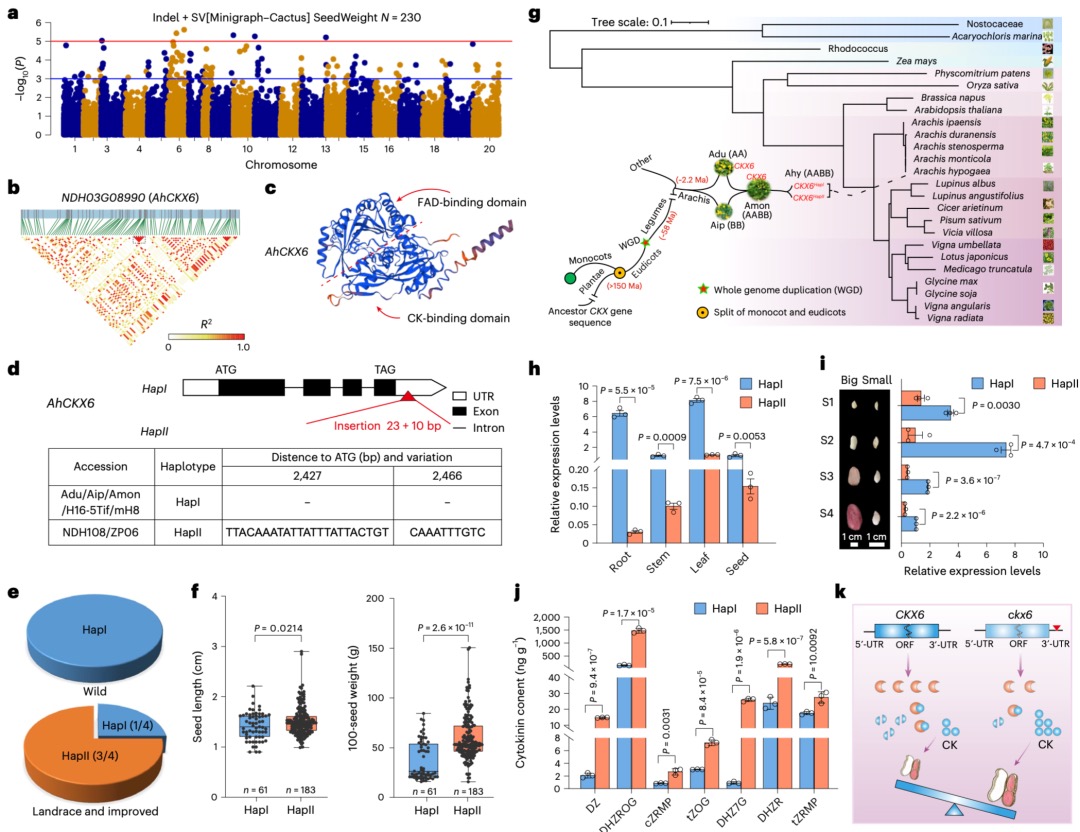
本研究发现,在蝗虫的体细胞(如大脑和腿肌)中,存在完全的剂量补偿效应,即雄性(X0)单条X染色体的基因表达水平通过特定的表观遗传调控(如H4K16ac富集和H4K20me1缺失)得以提升,使其与雌性(XX)两条X染色体的总表达量以及常染色体基因的表达水平相当;同时,研究通过转录组分析证实,雌性蝗虫的X连锁基因表达并非通过随机失活一条X染色体(如哺乳动物的X染色体失活),而是通过双等位基因同时表达来实现的,并且这一结论在排除A-to-I RNA编辑的潜在干扰后依然成立。这揭示了蝗虫在其大基因组背景下,采用了一种依赖于独特组蛋白修饰的剂量补偿机制以及双等位基因表达模式来协调性染色体基因剂量差异。
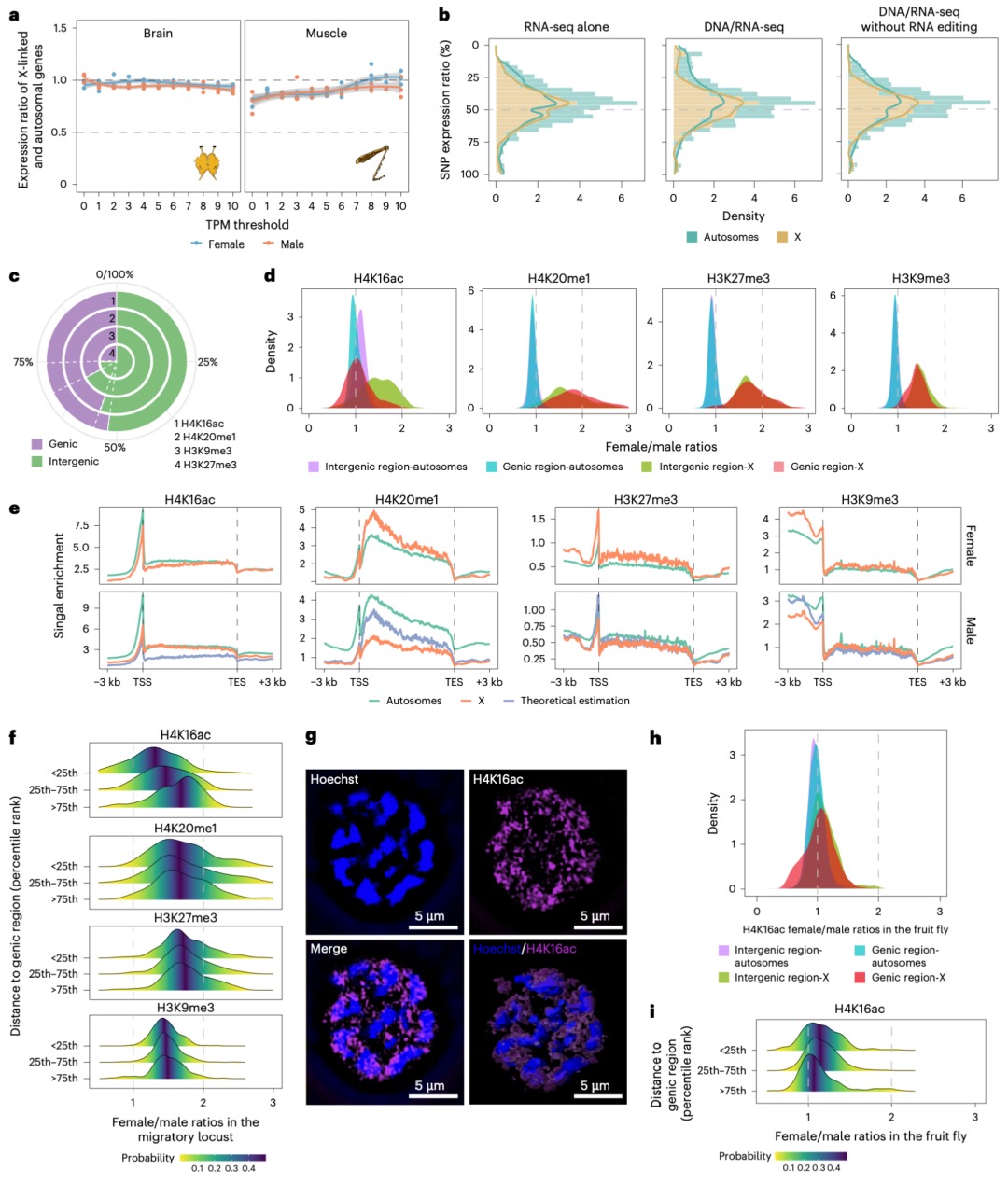
图5 体细胞中X染色体的平衡表达
本研究发现,在飞蝗的演化过程中,其X染色体通过广泛的染色体重排和基因易位形成。研究比较了飞蝗、沙漠蝗、菱蝗和蚤蝼的基因组,并将X连锁基因分为不同演化时期的类别。关键发现在于,近期才从常染色体易位到X染色体上的基因,其剂量补偿机制的建立存在滞后性:表现为抑制性组蛋白修饰H4K20me1的缺失不充分,而激活型修饰H4K16ac的富集程度又低于古老的X连锁基因,导致其剂量补偿不完全。相比之下,古老的常转X基因经历了更充分的适应性进化(dN/dS比率更高)并积累了更丰富的组蛋白修饰。研究还指出,性拮抗选择是推动某些对雌性有益(可能对雄性有害)的基因向X染色体易位的重要动力,这有助于在精子发生过程中通过减数分裂性染色体失活来抑制这些基因,从而提高雄性适应度。总之,该研究揭示了X染色体上新易位基因的剂量补偿机制需要时间逐步建立和完善,展现了剂量补偿系统的动态演化过程。
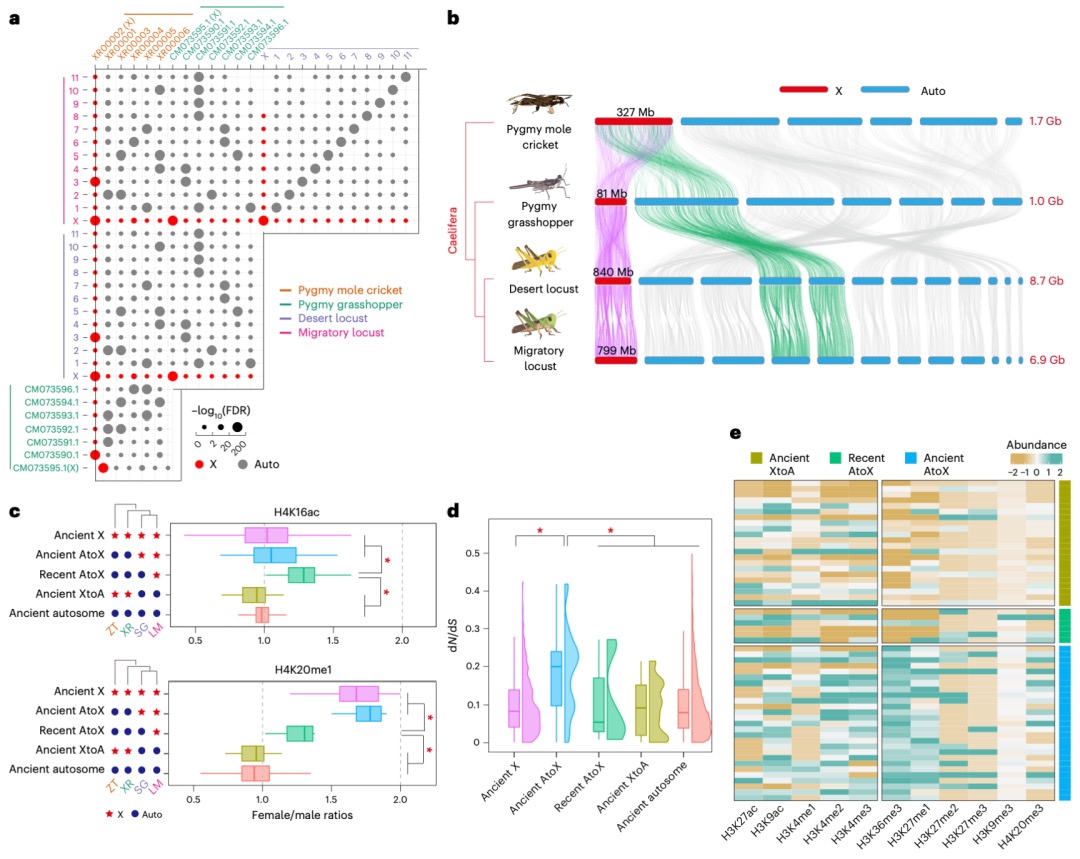
图6 源自常染色体的X连锁基因的剂量补偿转换
本研究基于基于高质量基因组的表观基因组图谱,深入揭示了蝗虫大基因组非编码区域(“暗物质”)的独特染色质调控特征。主要发现包括:1)基因组扩张伴随内含子区域增强子数量增加,其中增强子EH1被证实是调控蝗虫群居与散居行为可塑性的关键元件,且可能为蝗虫所特有;2)蝗虫在X0性别决定系统中演化出一种距离依赖性的剂量补偿机制,即通过H4K16ac的富集和H4K20me1的缺失来维持雄性体细胞X染色体表达平衡,但其效应在基因间区随距离增加而减弱,形成独特的嵌合式修饰模式,这与果蝇中均匀分布的模式显著不同;3)近期易位至X染色体的基因其剂量补偿机制的建立存在进化滞后性,表现为H4K16ac富集不足和H4K20me1缺失不充分,暗示大基因组中表观修饰信号的扩散效率受限,剂量补偿的重定位需要时间逐步完成。这些发现凸显了广阔的非编码区域为大基因组提供了独特的调控景观,其动态的表观遗传修饰是复杂生命现象的重要基础。