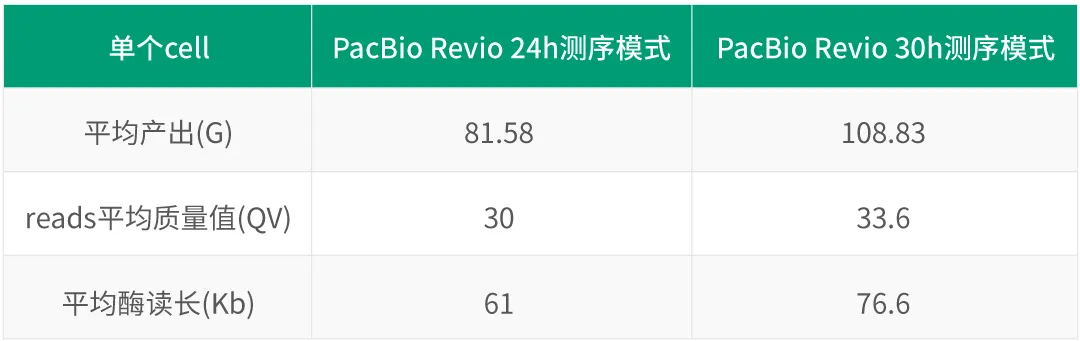
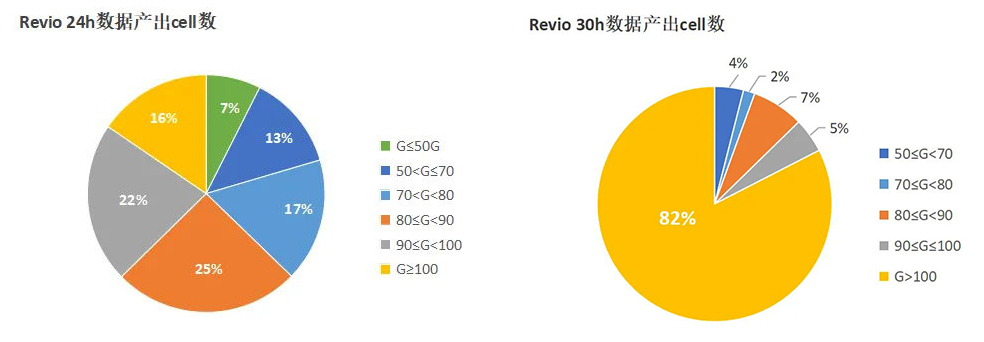
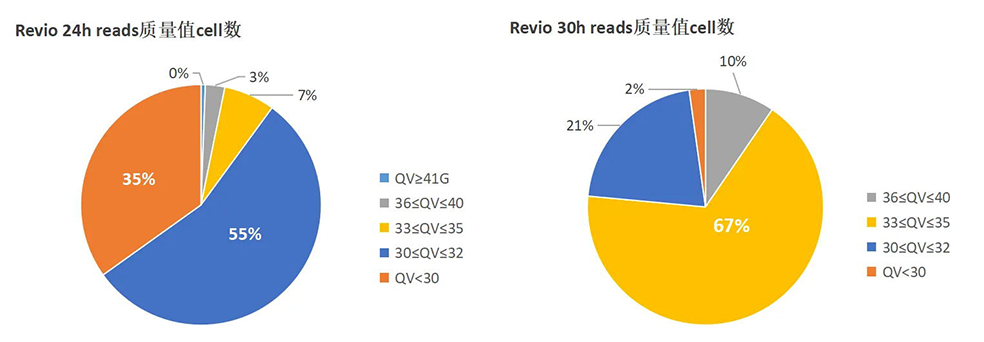
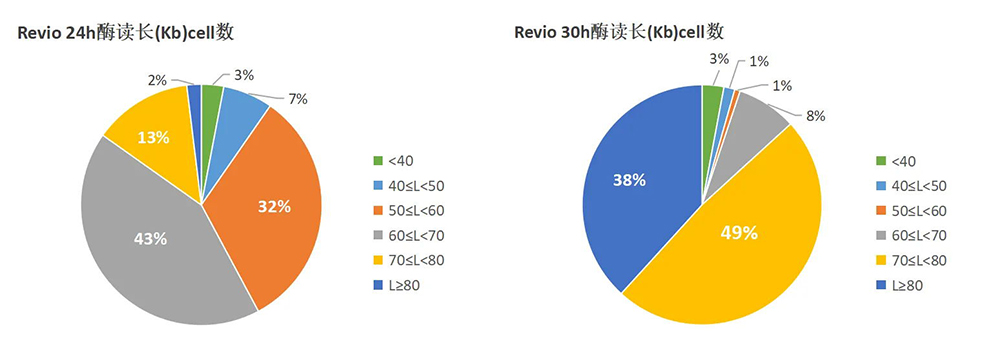
Nature Genetics | 朱玉贤院士团队发布首个棉花基因组完整图谱,阐述棉族独特折叠胚胎形成的分子与演化机制
植物种子及其周围结构提供的营养维持了人类文明的延续与发展。植物种子胚胎是营养的储存器,展现出丰富的结构多样性,反映了植物在进化过程中对环境适应的独特策略。1946年,早期植物学家A. C. Martin根据种子胚胎大小和形态特征,将植物种子胚胎划分为10种类型 (Martin, 1946),其中被子植物的胚胎通常表现为叶轴型(Foliate axile types, FA),包含了四种基本类型,即Spatulate(FA1)、Bent(FA2)、Folding(FA3)和Investing(FA4)。棉花(锦葵科植物)作为全球最重要的经济作物之一,具有复杂折叠的叶轴型胚胎,一般情况下,其子叶通过多层折叠完全包裹胚轴和胚根。与锦葵科近缘物种木槿相比,棉花显然经历了种子胚胎形态革新,从简单折叠胚(FA3)演变成复杂折叠胚类型(complex FA3),这种复杂折叠胚胎被认为是被子植物中发育最完全、最复杂胚胎类型(图1)。胚胎复杂折叠不仅能够保护胚根和胚轴,而且种子变大,能在有限种子空间内包裹最多的子叶从而提升储存营养资源的容量。同时,这一结构还与种子萌发、休眠及对环境的适应性密切相关 (Fryxell, 1978)。然而,棉花复杂折叠胚胎的发育过程及其背后的分子机制尚未被研究。
自朱玉贤院士团队与合作者在2012年首次公布雷蒙德氏棉基因组以来,棉花基因组学取得了一系列重要进展,推动了功能基因组学研究以及棉花复杂性状的解析 (Du et al., 2018; Huang et al., 2021; Huang et al., 2020; Wang et al., 2012)。然而,棉花基因组的准确与完整解析,尤其是复杂的转座子序列及其生物学功能,尚需深入研究与探讨。
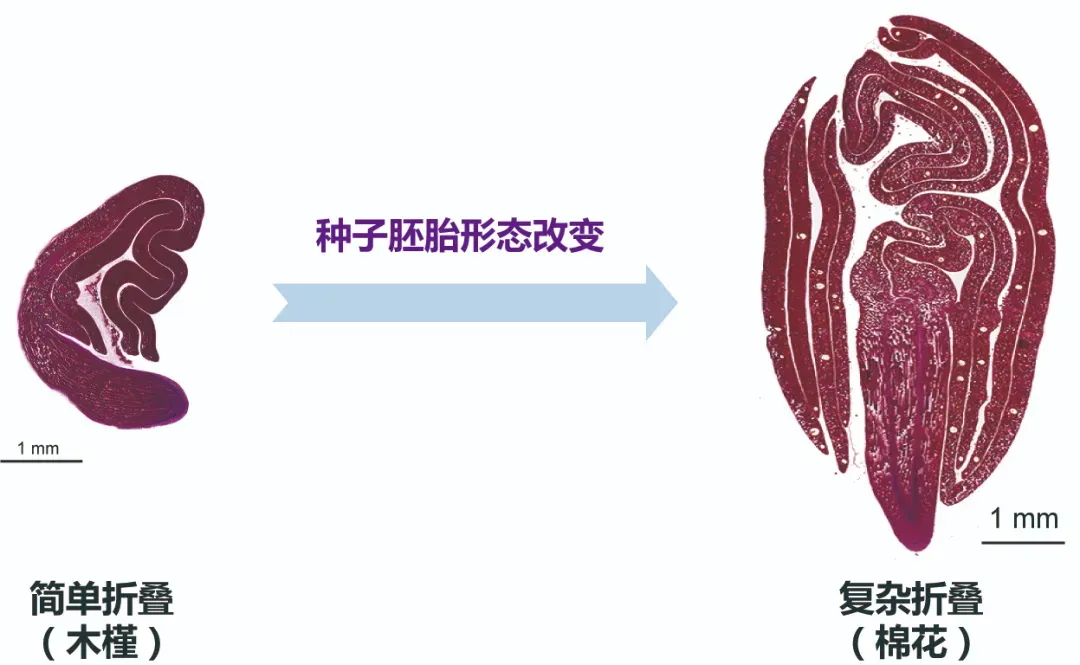
图1 棉花通过种子胚胎形态革新产生复杂折叠胚胎
2024年8月15日,武汉大学/北京大学教授朱玉贤,北京大学博士后黄盖(现为中国科学院遗传与发育生物学研究所副研究员)为主要作者在国际知名期刊Nature Genetics发表题为A telomere-to-telomere cotton genome assembly reveals centromere evolution and a Mutator transposon-linked module regulating embryo development的研究论文。该研究通过解析首个端粒到端粒的雷蒙德氏棉(Gossypium raimondii,四倍体棉的祖先种)基因组完整序列图谱,揭示了其独特的着丝粒结构类型及表观图谱。通过深入挖掘功能性转座子,发现由三个新分子(miR2947-DNA转座子MuTC01-加倍基因LEC2b)组成的三级小RNA调控机制,从而阐明了棉花复杂折叠胚胎形成的分子调控与演化机制 (Huang et al., 2024)。

图2 朱玉贤院士团队在棉花基因组和功能研究取得重要进展
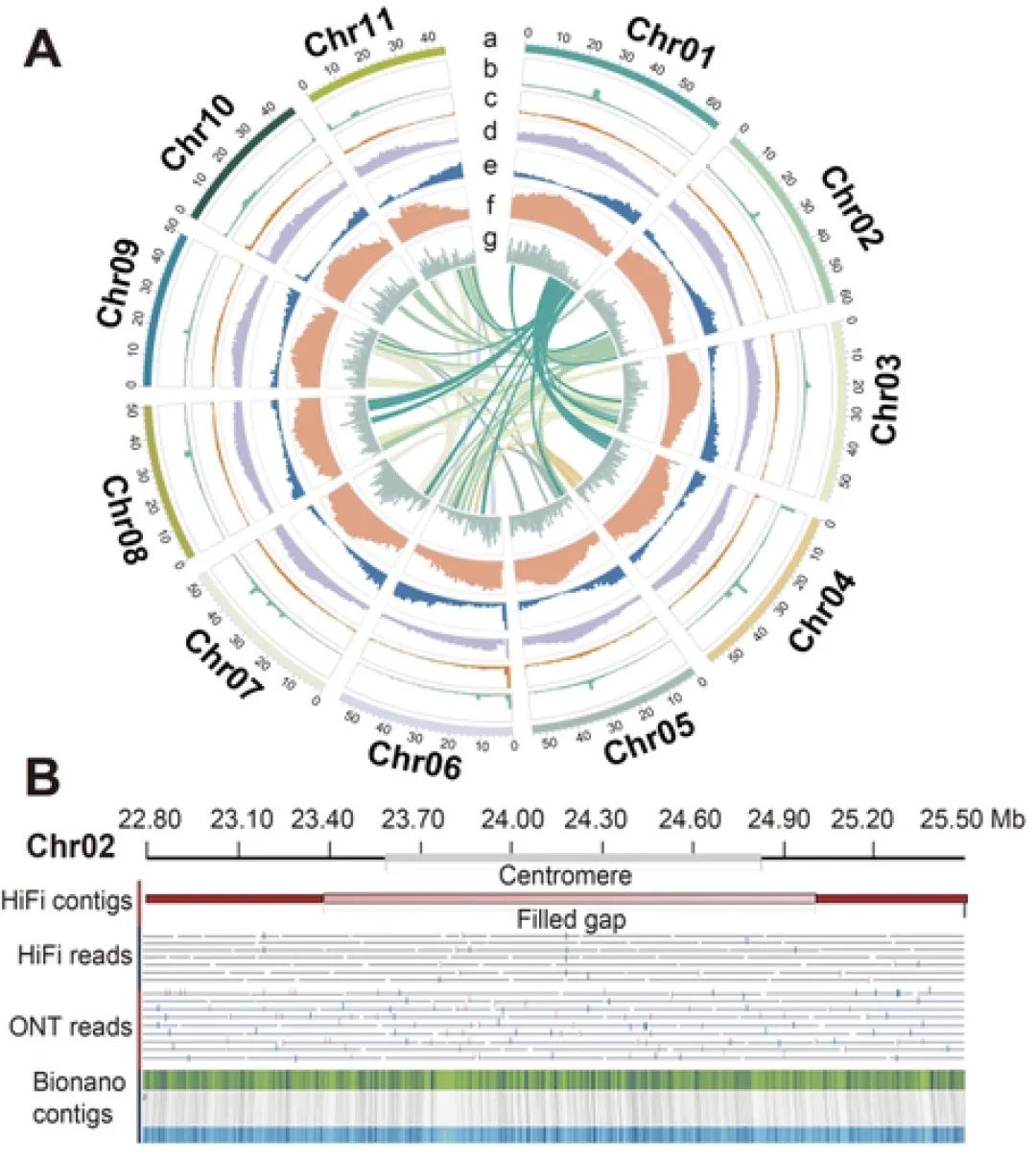
该研究整合了最新的测序技术和算法(希望组为本研究提供了NGS、超长和HiFi测序。),成功获得了776 Mb首个二倍体棉花基因组完整序列图谱。与以往基因组版本相比,首个棉花基因组完整序列图谱具有高连续性和完整性,成功组装了着丝粒和端粒序列,并对转座子和基因进行了更精确和完整的注释,识别出53167个蛋白质编码基因,显著高于以往版本(37505–40976个基因)。此外,T2T基因组还修正了之前版本中的错误序列,主要是涉及着丝粒、端粒等复杂区域。通过深入解析着丝粒序列,发现了雷蒙德氏棉着丝粒独特的结构与组成(图3)。雷蒙德氏棉着丝粒主要由LTR类转座子构成,缺乏短着丝粒微卫星序列,展现出与其他植物显著不同的特征。此外,雷蒙德氏棉的着丝粒缺乏典型的核小体有相位的排布规律,这一差异主要源于其着丝粒的形成过程直接受到长末端重复逆转录转座子入侵的影响。

图3 雷蒙德氏棉基因组具有独特的着丝粒结构
基于基因组完整序列图谱,研究者对棉花转座子进行精准鉴定,得到了872549条非冗余转座子序列。棉花含有丰富的TIR类转座子,其中Mutator家族是最主要的TIR类型。转座子元件表达分析发现,只有约2%的序列编码了具有转录活性的转座子,而在棉花胚胎发育晚期有88个转座子在子叶阶段表现出组织特异性表达特性(图4)。这些具有组织特异活性转座子中,仅DNA MuDR转座子(命名为MuTC01)能够产生最丰富的正负链小RNA,是反式作用siRNA产生位点。分析发现,MuTC01起源于DNA转座子Mutator家族,在全基因组中具有34个同源拷贝,只有MuTC01能产生高丰度的siRNA,预示MuTC01可能通过siRNA在棉花胚胎发育过程中发挥作用。
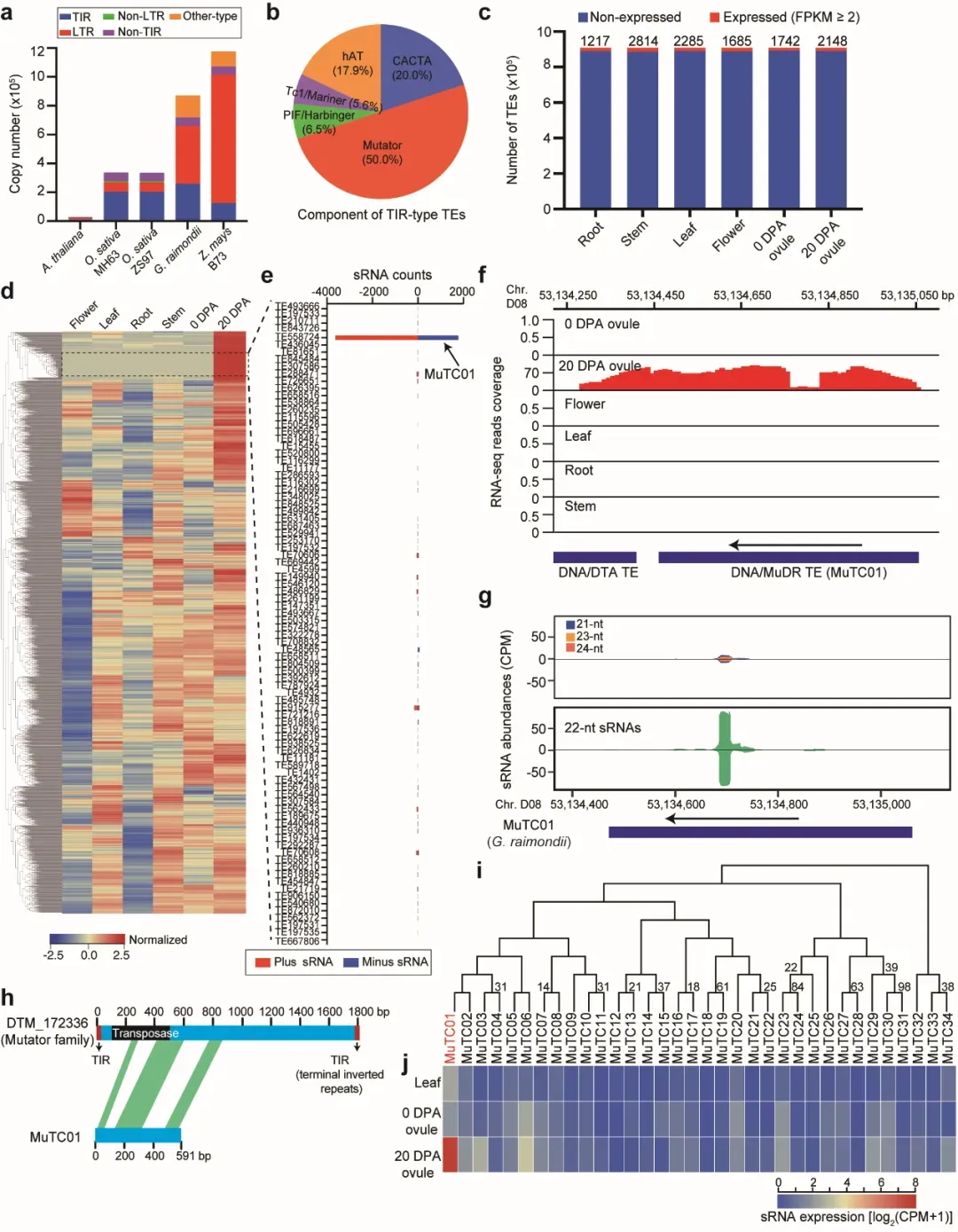
图4 转座子功能分析揭示了胚珠特异表达并产生siRNA的MuTC01转座子
通过靶向预测以及降解组分析,他们发现MuTC01受棉花特异的miR2947靶向切割产生有相位的siRNA(图5)。进一步通过CRISPR–Cas9基因编辑技术,对棉花的miR2947和MuTC01进行基因突变实验。电镜观察成熟胚胎形态显示,突变体mir2947和mutc01都表现出胚胎发育异常表型,子叶没有被完整包裹和折叠。
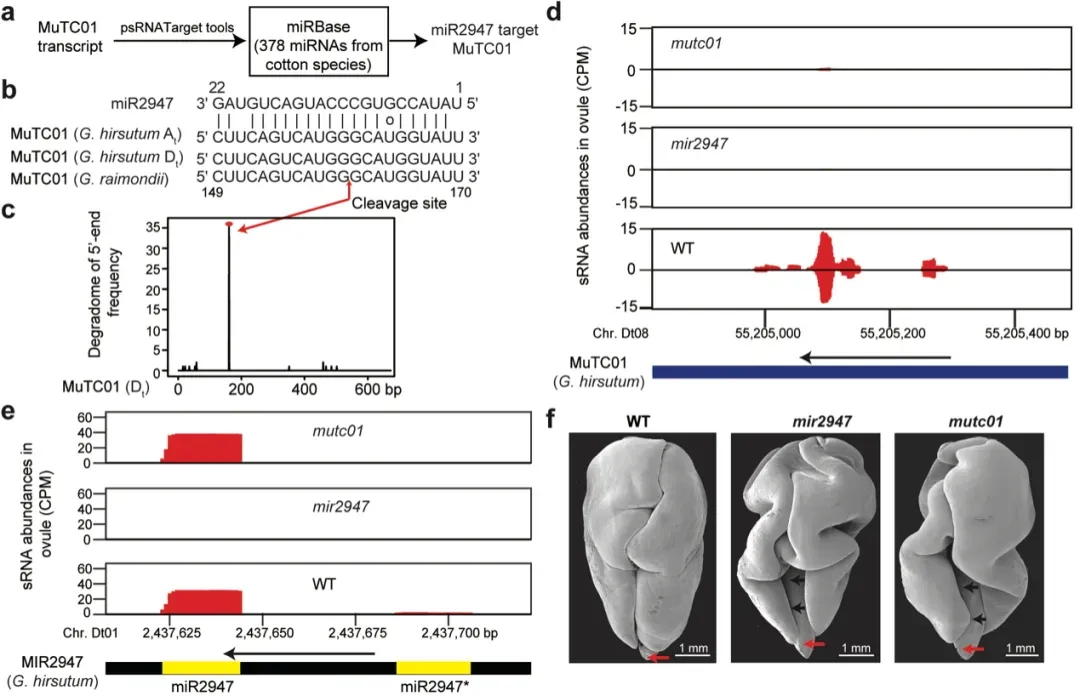
图5 miR2947靶向MuTC01产生小RNA调控棉花胚胎折叠
通过对棉花胚胎发育轨迹进行切片观察(图6),显示棉花突变体 mutc01和mir2947均会导致胚胎折叠异常的表型,特别是在胚胎发育后期(开花后23天以后)变得尤为明显。这些结果表明 miR2947–MuTC01调控模块在棉花胚胎发育中起到关键调控作用,突变体胚胎形态与近缘种木槿相似,表明miR2947–MuTC01 调控模块很可能是棉花胚胎复杂折叠类型形成的关键因素。
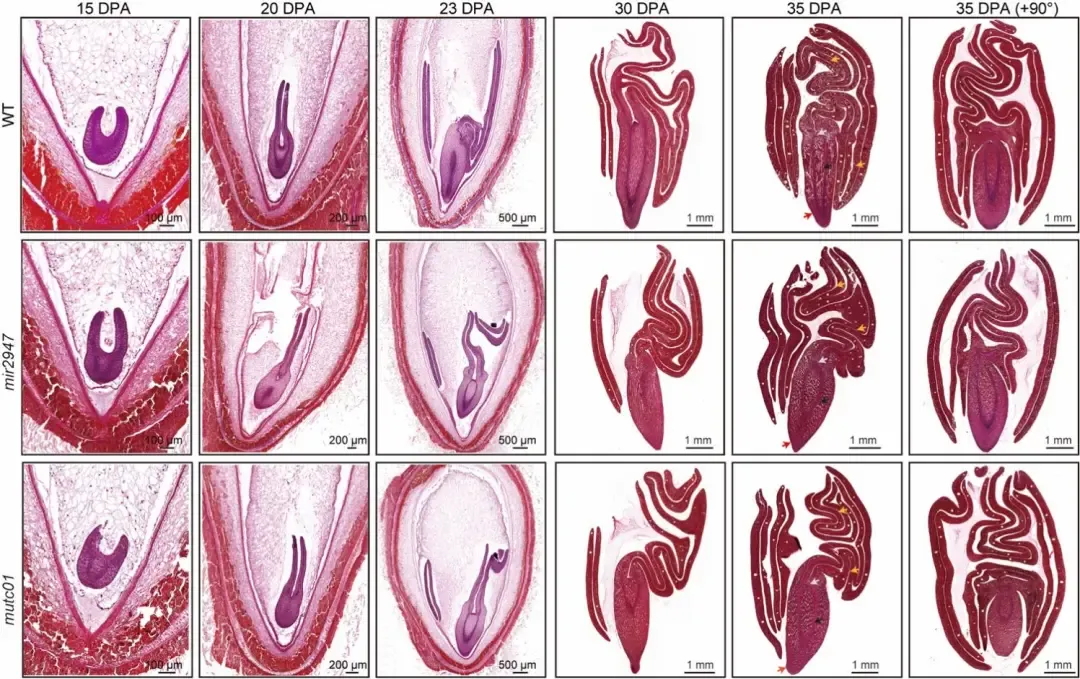
图6 棉花突变体胚胎发育轨迹切片观察
为进一步探究miR2947–MuTC01调控模块下游的靶标,他们结合靶位点分析、转录分析以及切割位点验证等实验,确定MuTC01产生的22-nt siRNA(命名为siRNA_22nt)能够靶向棉花LEC2b基因(图7)。系统演化分析发现,LEC2基因起源于棉属全基因组加倍事件,在棉花中有两个拷贝,分别命名为LEC2a和LEC2b。与拟南芥、可可同源的拷贝为LEC2a,棉花独特的基因为LEC2b。LEC2a和LEC2b在第一个外显子区域存在553 bp的变异区域,使得MuTC01能靶向LEC2b产生21-nt有相位的siRNA,而不能靶向LEC2a。两个同源基因独特的序列和调控演化暗示LEC2a和LEC2b存在功能分化。
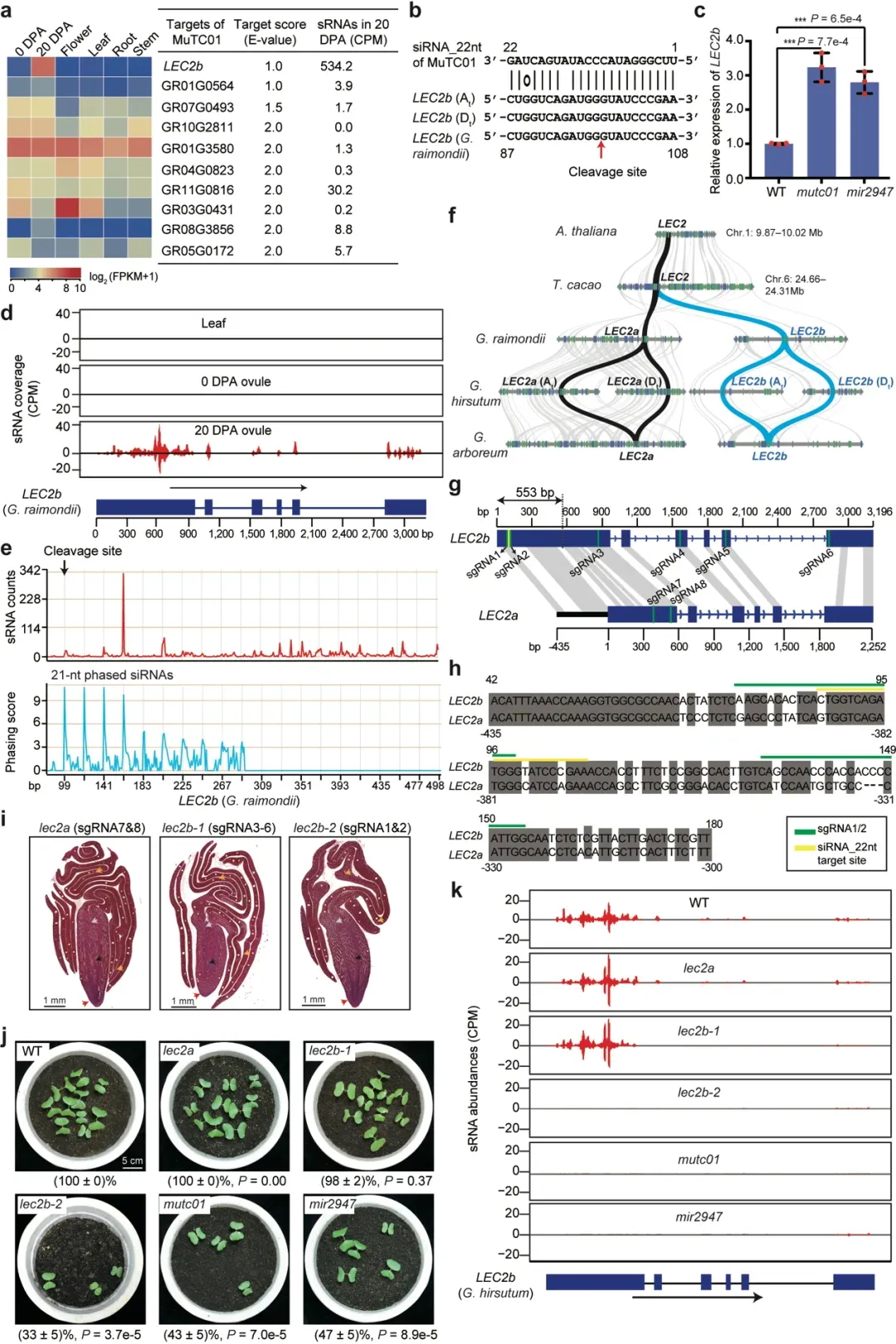
图7 由miR2947-MuTC01-LEC2b组成的三分子模块调控棉花胚胎折叠
作者进一步利用基因编辑实验创造了三个棉花突变体(图7),包括:在LEC2b的第一个外显子区域设计两个sgRNA,编辑siRNA_22nt 靶向LEC2b的区域,获得棉花突变体lec2b-2;在LEC2b外显子设计四个sgRNA,编辑LEC2b蛋白质编码区,而不编辑siRNA_22nt靶向区域,获得棉花突变体lec2b-1;在LEC2a设计两个sgRNA,编辑LEC2a蛋白质编码区,获得棉花突变体lec2a。他们通过棉花胚胎的发育轨迹进行切片观察,发现棉花突变体lec2a和lec2b-1在棉花胚胎发育过程中无明显的发育异常表型,而lec2b-2突变体子叶不能正确包裹胚胎,类似于mutc01和mir2947等棉花突变体,且在胚胎发育后期(开花后23天以后)变得尤为明显。
作者进一步检测五个棉花突变体(mir2947, mutc01, lec2a, lec2b-1, lec2b-2)在LEC2b基因位点的siRNA表达水平(图7)。数据表明,在mir2947, mutc01,lec2b-2棉花突变体背景下,LEC2b基因位点有相位的siRNA消失,而在lec2a和lec2b-1突变体背景下,不影响LEC2b基因位点的siRNA的产生。这个siRNA分布情况与突变体的表型完全一致。这些数据表明,miR2947–MuTC01–LEC2b三分子模块是通过LEC2b产生三级siRNA控制棉花胚胎复杂折叠,而不是通过影响LEC2b蛋白质功能而发挥作用。
作者进一步探究miR2947–MuTC01–LEC2b三分子模块的起源与演化(图8),结果表明该三分子模块同时存在于具有复杂折叠胚胎类型的整个棉族(包括棉属在内的100多个种),显著不同于其近缘物种木槿族所具有的简单折叠胚胎类型。因此,作者提出了三级小RNA调控棉族独特胚胎类型的分子和演化机制,即棉族特异的MIR2947产生第一级22-nt的miR2947,直接靶向DNA转座子MuTC01,产生第二级小RNA,再靶向全基因组加倍产生的LEC2b基因,产生第三级小RNA,从而调控棉族复杂折叠胚胎形成(图8)。这项研究系首次在植物界发现具有功能的三级小RNA调控机制,也是首次从发育角度阐释棉族复杂胚胎折叠过程以及背后的分子与演化机制。
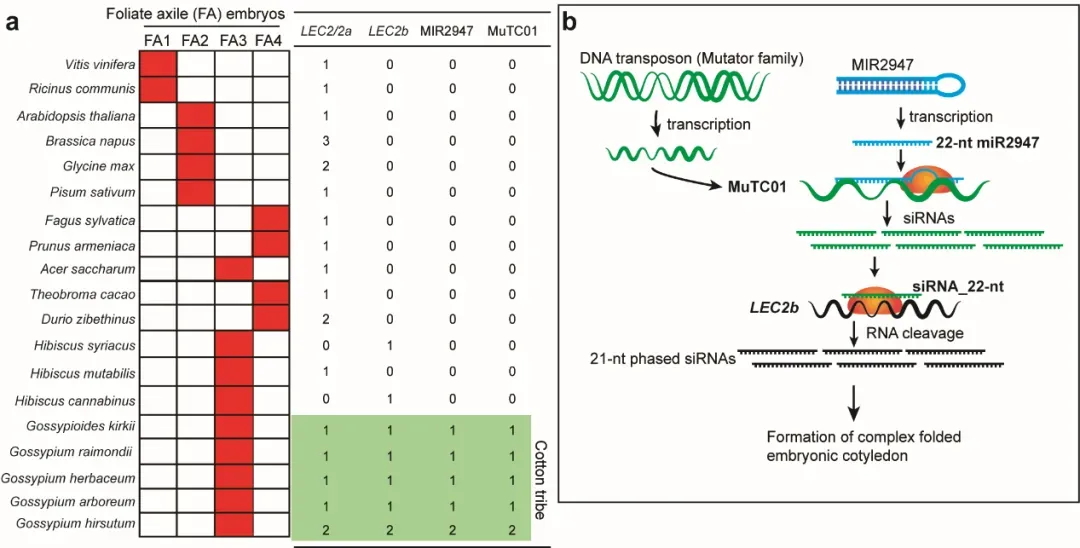
图8 棉族复杂折叠胚胎形成的分子和演化机制


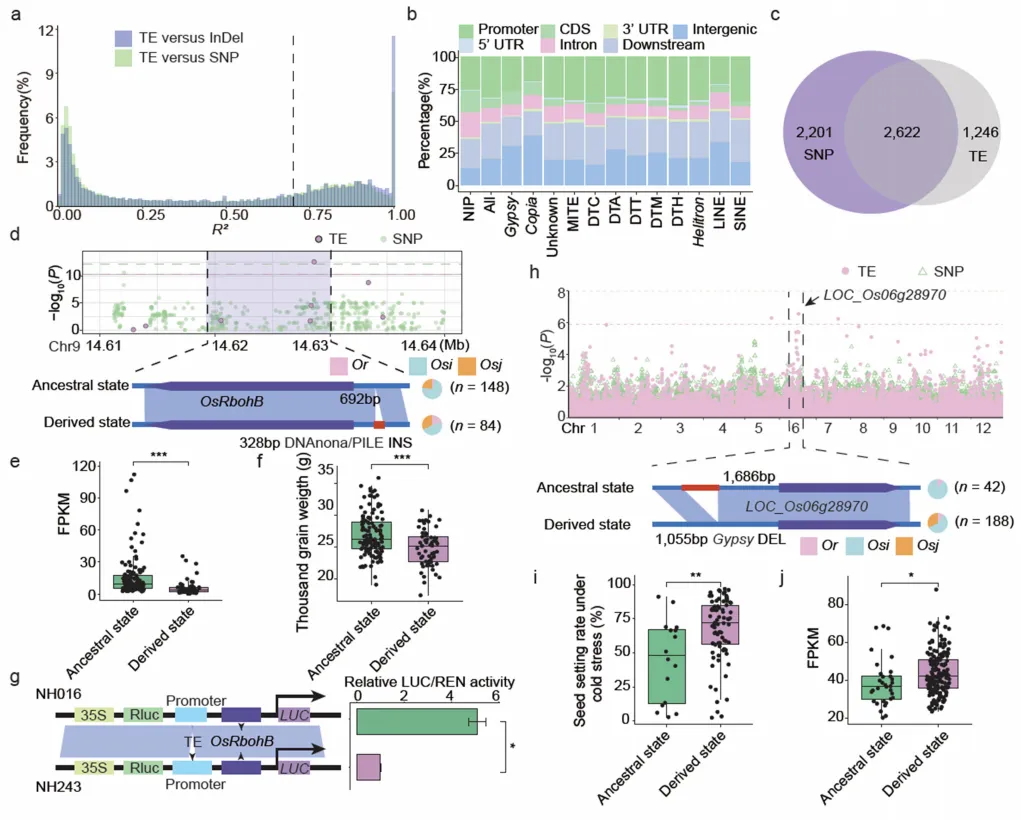 图3 TE变异影响水稻基因表达和农艺性状
图3 TE变异影响水稻基因表达和农艺性状