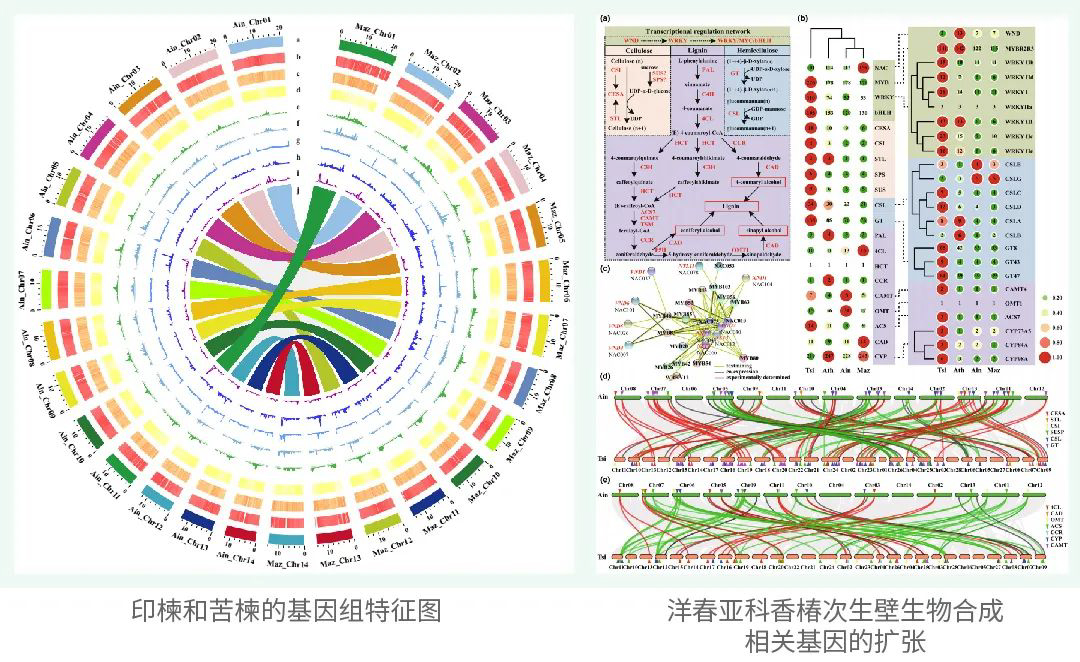
署名文章 | Nature Genetics!希望组携手赖锦盛教授团队再创新里程—大型真核生物玉米T2T无间隙基因组组装
2023年6月15日,中国农业大学国家玉米改良中心、玉米生物育种全国重点实验室赖锦盛教授团队以题为“A complete telomere-to-telomere assembly of the maize genome”在国际知名期刊Nature Genetics《自然·遗传学》上在线发表了玉米全基因组所有染色体端粒到端粒完整无间隙组装结果,在复杂动植物基因组中第一个实现真正意义上的全基因组完整无间隙组装。该研究是复杂基因组组装领域工程技术研究的重大突破,攻克了复杂动植物基因组组装的最后一道难题,是基因组组装和基因组学研究的一个重要里程碑。

赖锦盛教授为该论文通讯作者,中国农业大学陈建副教授、博士研究生王子健为该论文共同第一作者。中国农业大学金危危教授、宋伟彬教授、赵海铭副教授、辛蓓蓓副教授、黄伟老师、史俊鹏博士后(现已出站),爱荷华州立大学Matthew B. Hufford教授、内布拉斯加大学林肯分校James C. Schnable教授、中国科学院遗传与发育研究所韩方普研究员和刘阳博士,以及北京希望组生物科技有限公司为该研究提供了重要帮助,希望组员工(胡江、王超)有幸成为了共同作者。该研究得到了国家重点研发计划、国家自然科学基金、海南崖州湾种子实验室、崖州湾科技城管理局、河南省科技厅以及河南现代种业有限公司的资助。希望组为本研究提供了HiFi、ONT超长测序、NextDenovo(v2.2-beta.0)初步组装和NextPolish(v1.1.0)基因组矫正服务。
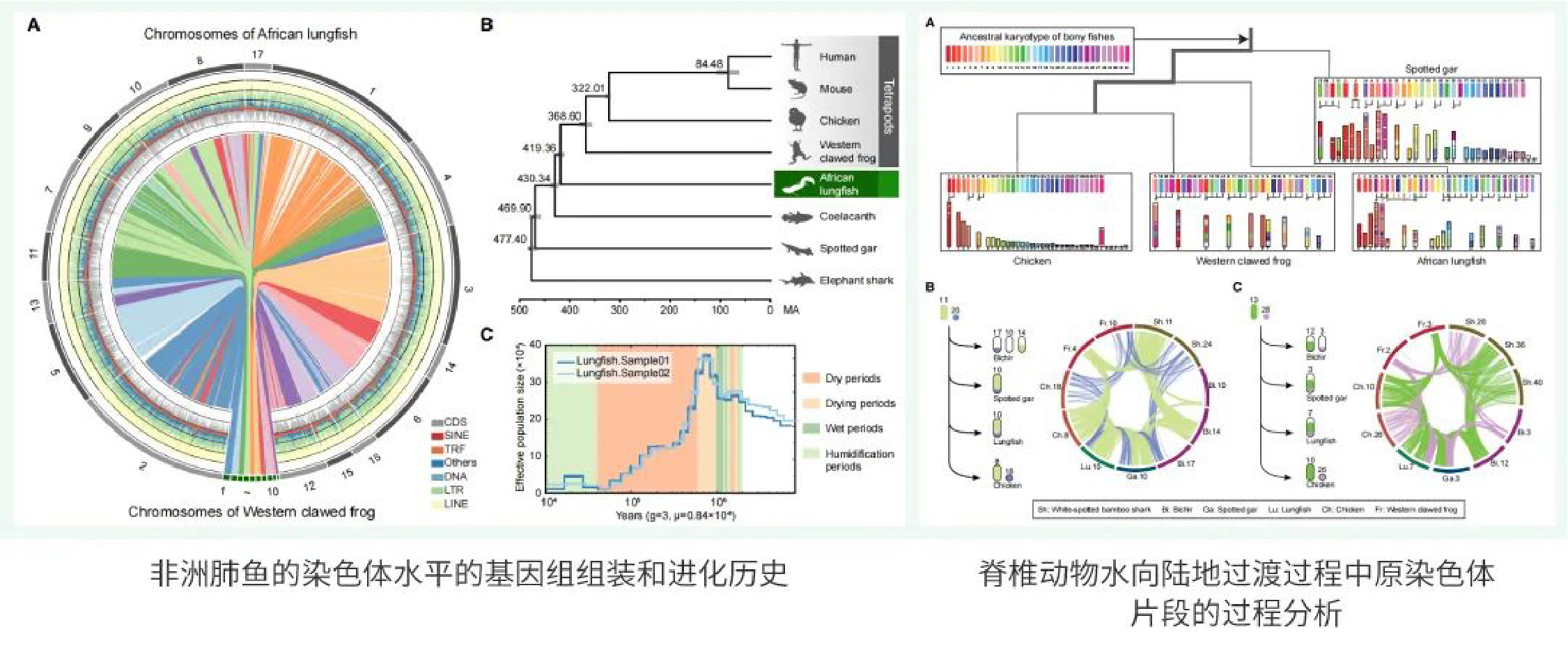
玉米是世界范围内的重要作物,其基因组组装对玉米基础研究和分子育种均有重要意义。同时,玉米也是经典的复杂基因组研究的模式植物。自2009年玉米基因组草图公布以来,已有近50个不同玉米自交系基因组被组装。然而,由于玉米基因组大(与人类基因组相近),且拥有超过80%的重复序列,目前已报道的玉米基因组都存在数百或数千个“空白”区域未被解析。
Mo17自交系是经典的玉米杂种优势群Lancaster群的代表。Mo17自交系及其衍生材料在我国玉米生产中被广泛应用。赖锦盛教授团队以Mo17自交系为材料,综合利用了约237×的ONT Ultra-long和约69.4×的PacBio HiFi测序数据,完成了最新的玉米基因组组装,其大小为2,178.6 Mb,每条染色体的端粒到端粒均由一条完整连续的序列组成,碱基精确度超过99.99%。最新的组装不仅在过去高质量组装的基础上增加了1029个基因,还解锁了玉米基因组中结构最为复杂、从未被组装的基因组空白区。这是首个完整的、无间隙的玉米基因组序列,也是首个所有染色体都完整组装的复杂动植物基因组。
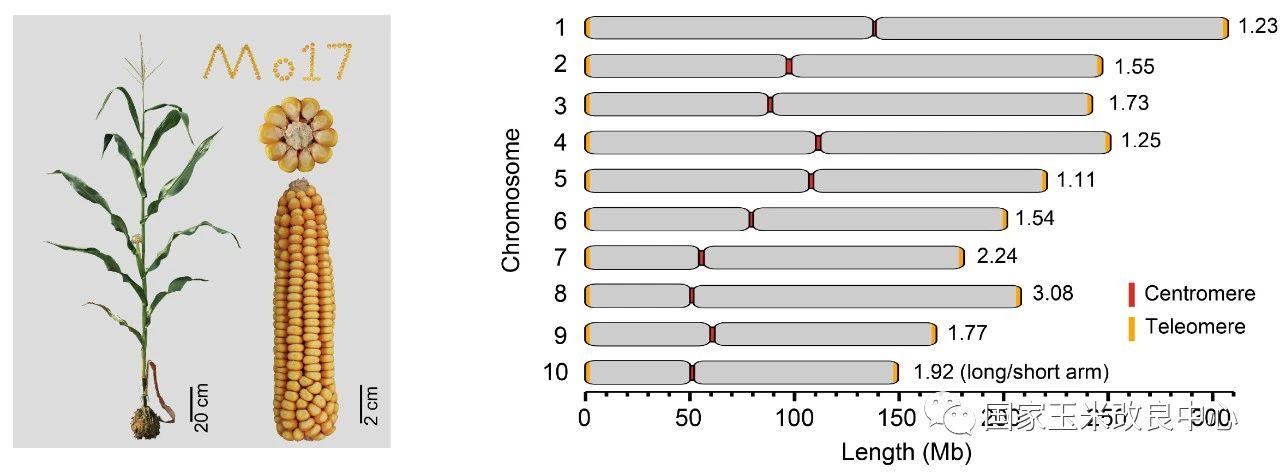
图1. Mo17基因组完整组装
最后,作为专门的安排,赖锦盛教授在致谢部分特别标注,用该研究成果的发表来纪念他的博士后研究阶段的导师Joachim Messing (1946—2019)。 Messing教授是国际上分子生物学先驱和“鸟枪法”测序技术的倡导发明者,是美国科学院和德国科学院院士,于2013年获得沃尔夫(Wolf)奖。
本文转载自:国家玉米改良中心
项目文章 | 希望组再次参与Nature论文工作—中国人群泛基因组
由复旦大学、西安交大、中国医学科学院等26家单位联合发布的中国人群泛基因组联盟(CPC)一期研究进展,初步构建了首个中国人群专属的泛基因组参考图谱,为破译中国人群基因密码奠定基础,为“健康中国”“精准医学”战略提供支撑。
6月14日,相关成果以《基于36个族群的中国人泛基因组参考图谱》(“A Pangenome Reference of 36 Chinese populations”)为题发表于《自然》(Nature)主刊。这也是我国学者领导的人群基因组研究首次发表在《自然》主刊。复旦大学徐书华教授、西安交通大学叶凯教授、中国医学科学院褚嘉祐教授和复旦大学陆艳副教授为该文的共同通讯作者,西安交通大学杨晓飞副教授、复旦大学博士后高扬、中国科学院上海营养与健康研究所博士生陈豪、谭昕江、中国医学科学院杨昭庆研究员、复旦大学邓恋青年研究员为论文的并列第一作者。研究工作得到了国家自然科学基金、科技部重点研发计划等项目的资助。希望组为本研究提供了部分样本的HiFi、ONT、Hi-C、Bionano和Iso-seq测序服务。
“基因组结构变异大概是生物进化中从微观到宏观演变的关键遗传基础,也是最有可能连接渐变到跃变这个‘鸿沟’的进化密码。”复旦大学校长、中科院院士金力点评,“我相信通过对基因组结构变异的高精度解析,不但能大幅提升‘基因型-表型’关联分析的功效,而且有可能最终帮助我们理解生命演化中重要性状和功能产生的遗传基础和分子机制。”
独立自主完成首个中国人群泛基因组参考图谱
作为人口大国,我国巨大的人口基数和丰富的人群多样性是发展人类基因组学和精准医学的重要优势:西南部高原地区分布着众多藏缅、南亚语系族群,东西方人群在西北部丝绸之路沿线交融,苗瑶语族人群在云贵地区世代繁衍,蒙古、突厥人群曾游牧于北部风沙地,通古斯语族抵抗严寒一路向北,台-卡岱(侗台)族群的先辈亦曾穿梭于南方丛林河谷。悠久的人群历史、丰富的地理气候环境,塑造了中华民族独特的遗传多样性,构成了人类泛基因组研究不可或缺的东方画卷。构建能够代表中华民族遗传多样性的中国人群泛基因组图谱势在必行且迫在眉睫,这将极大提高捕获罕见或低频遗传变异的灵敏度和准确性,支撑服务中国人遗传多样性研究、复杂疾病分子机制研究和精准医学研究与应用。

中国人群泛基因组联盟”一期36个族群画像集
中科院院士、分子微生物学家赵国屏认为:“这一成果表明我国科学家在人类基因组学领域的研究水平得到了显著提升。我相信这项工作对我国的人类基因组学和医学中的复杂疾病遗传基础研究等领域会起到重要的推动作用。”
助力遗传学、医学研究,服务人民生命健康
在第一期研究计划中,CPC对代表中国36个族群的58个样本采用最新的第三代高保真基因组测序技术进行了深度测序,结合最新的单倍型基因组组装方法,获取了116个高质量单倍型基因组,并以图基因组的方式构建了高质量中国人群参考泛基因组。该泛基因组图谱总共包含约3.01 Gb个碱基对的序列信息,在现有人类参考基因组的基础上新增了约1.9亿个碱基对的新序列,包含约590万个小变异(单核苷酸多态性变异和小规模插入/缺失变异)和约3.4万个结构变异(Structural variation, SV),涉及至少1367个蛋白编码基因复制事件等。其中,约500万个碱基对新序列存在于95%以上的单倍型中,被视为中国人群基因组核心序列,可能与中国人群特有的较为稳定的生物学功能或表型特征相关。
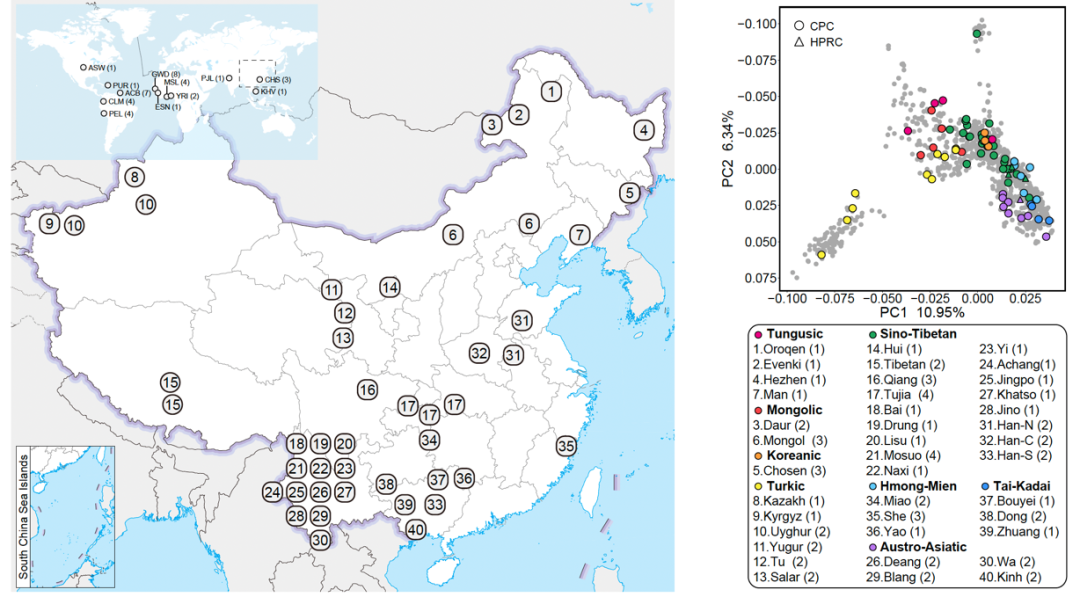
CPC一期核心样本地理分布及语系、族群、遗传聚类关系
同时,CPC新发现的遗传变异影响了具有潜在功能和经受过适应性进化的基因,这些基因可能与亚洲人群特有的疾病易感性及表型多样性有关,这也证实了将人群专属高质量泛基因组用于基因组学和医学研究的潜力和必要性。此外,研究人员在CPC参考图谱中发现了相当大比例的古人来源基因序列——平均每个族群和每个样本中分别有约15Mb和约9.5Mb的古人来源新序列——这可能是前期开展大量研究却未在现代人基因组中发现的古人基因渗入序列,或将为东亚现代人基因组中的古人基因渗入研究乃至整个古DNA领域提供新的信息资料和线索。
未来,中国人群参考泛基因组不仅有助于中华民族共同体的遗传学研究,还将改变过去依赖主体基于欧洲白人的参考基因组而导致东亚特有罕见变异检出精确度难以提升的困境,应用于我国重大疾病的遗传机制解析,从而提高我国生物医学数据分析的质量和效率,服务人民生命健康。
中国人群泛基因组图谱已公开在线发布:
https://pog.fudan.edu.cn/cpc/#/;https://github.com/Shuhua-Group/Chinese-Pangenome-Consortium-Phase-I
该项研究所涉及的样本信息和数据的公开发表已获得国家人类遗传资源管理部门批准。该项研究得到了国家自然科学基金重点项目、基础科学中心、国家重点研发计划等项目的资助。
论文链接:https://www.nature.com/articles/s41586-023-06173-7
原文转载自:复旦大学
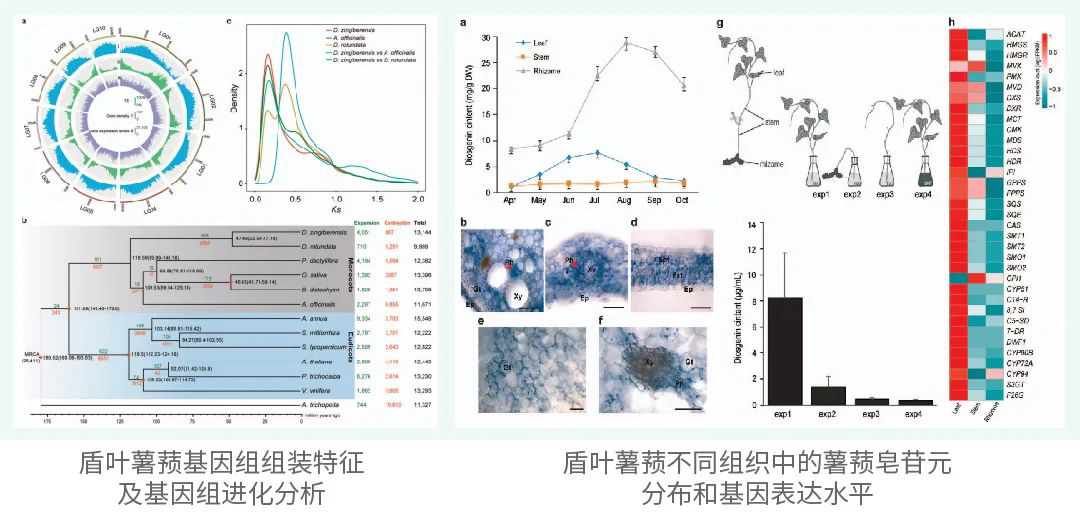
署名文章|Science! 中国科学院昆明动物研究所在灵长类进化遗传领域取得重大突破
人类长期关注灵长类动物的起源和演化。该方向研究不仅有助于回答人类起源问题,也有助于我们更多地了解人类独特身体结构特征的演变历史。非人灵长类动物在生物学、演化学、药理学等领域中扮演着重要角色,但目前仅有不到10%的非人灵长类动物的参考基因组被测序。
浙江大学生命演化研究中心张国捷教授团队联合昆明动物研究所吴东东教授团队、西北大学齐晓光教授团队和其他国内外合作者在Science杂志在线发表题为“Phylogenomic analyses provide insights into primate evolution”(基因组学分析提供了对灵长类演化的洞见)的研究论文。该论文回答了与灵长类演化相关的一系列问题。
灵长类动物演化或受物种大灭绝事件影响
据介绍,此次研究对象覆盖了50个灵长类动物物种,跨越了38个属和14个科,其中包括了之前研究中较少涉及的新世界猴和原猴;研究中有27个新的高质量基因组数据,这些新数据可以提供更多、更准确的遗传信息。全面的数据则有利于更深入地了解灵长类动物的演化历程。专家表示,这个时间距离6550万年前那次造成非鸟恐龙灭绝的白垩纪末期大灭绝事件非常近,大致临近白垩纪与古近纪交界时间。这意味着灵长类动物的演化可能受到了物种大灭绝事件的影响。
此外,研究人员通过重建灵长类的祖先核型演化过程,观察到在染色体水平上核型演化模式总体上是保守的。这表示在不同谱系之间,染色体大多数都保持了类似的结构和数量。
最新研究采用了更多染色体级别的原猴物种进行研究,弥补了之前由于数据不足而导致偏差的问题。这项研究发现,人的8号染色体对应到原猴的两条染色体上。因此,研究人员推测类人猿下目祖先以及所有灵长类祖先中的两条染色体在狭鼻类出现后融合成一条染色体,最终演变成人类8号染色体。这项研究校正了前人对人类8号染色体在灵长类物种中的起源历程的推断。
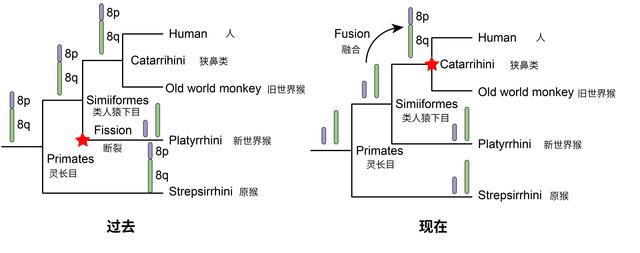
人类第8号染色体在灵长类起源过程中不同假说的示意图。张国捷课题组供图
灵长类动物大脑经历了快速演化
灵长类动物在演化过程中的大脑体积变化非常引人注目。最初的原猴亚目和眼镜猴,它们的脑容量非常有限,但随着时间的推移,新世界猴及旧世界猴出现后,它们的脑容量不断增大,最终在大猿类和人类的演化过程中,相对脑容量进一步增大。相对脑容量的增大与智力程度密切有关,同时也反映了物种演化适应环境的能力。
研究人员发现了一些基因在灵长类的演化历程中受到了强烈的正选择(即倾向于富集更多氨基酸变化)。这包括一些前人的实验研究已经发现的与大脑发育有关的关键基因,这些基因的突变会导致小鼠的大脑功能受损。此外,研究人员还发现了一些非编码区域在四个关键的灵长类演化节点(类人猿下目的祖先、狭鼻类祖先、大猿祖先和人类)中发生了加速演化。这些区域落在大脑发育相关基因的调控区域,这些结果表明了灵长类动物在漫长的演化过程中通过调节大脑相关基因的表达不断地优化大脑构造。
研究人员认为,这些发现表明在灵长类动物大脑演化成更发达形态的过程中,有很多基因和调控区域参与,这丰富了我们对灵长类大脑演化分子机制的认识。
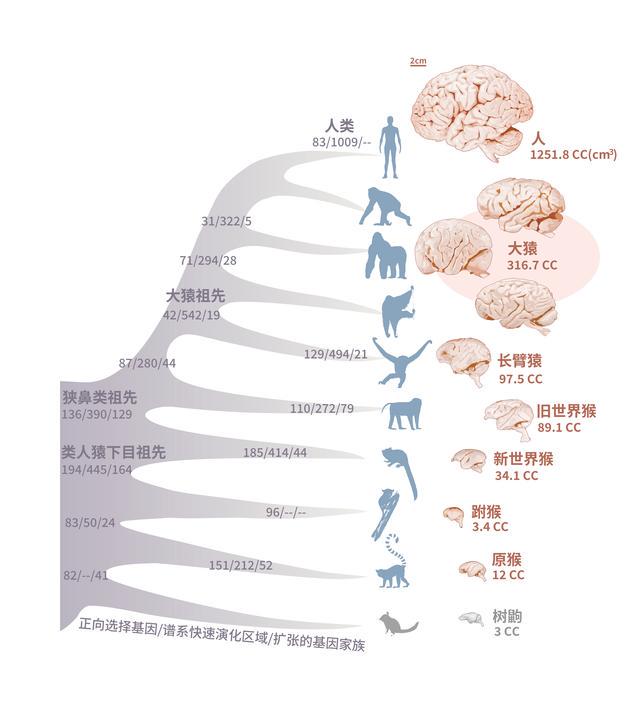
灵长类物种脑容量演化历程及此过程中基因组上的变化。张国捷课题组和吴东东课题组合作供图
猿类尾巴丢失或与基因调控序列突变有关
尾巴是很多动物的标志性特征之一,尤其是对于一些动作灵敏的灵长类物种,长短不一的尾巴能够帮助它们稳定身体、转向和控制速度。“然而,猿类和人都失去了尾巴,这成为它们区别于其他灵长类的重要特征。研究表明,这一现象可能与一些特定的基因调控序列的突变有关。”周龙说。
在人猿共同祖先中,研究人员检测到多个基因的非编码调控区域积累了大量变异,其中包括KIAA1217。人的KIAA1217基因发生突变可能会导致脊柱和尾椎畸形,影响脊柱的正常发育;而在小鼠中,这个基因的突变则会导致尾椎数量的减少。研究人员发现,这个调控区域落在基因的增强子区域,并且与KIAA1217基因落在同一个拓扑结构关联域中,证明这个区域和基因有很强的交互作用,可能调控了KIAA1217基因的表达。
不同的灵长类有不同的饮食习惯和消化系统,有些灵长类如叶食性的疣猴喜欢吃树叶。为了适应这种饮食,它们演化出独特的前肠系统。此次研究还发现了一些关键的消化基因在疣猴的祖先受到正选择而积累了特殊的氨基酸变异,来适应这种特殊饮食的状态。疣猴演化出能够消化脂肪酸的能力,配合它们独特的前肠和肠道微生物使它们能够应付食叶性饮食。
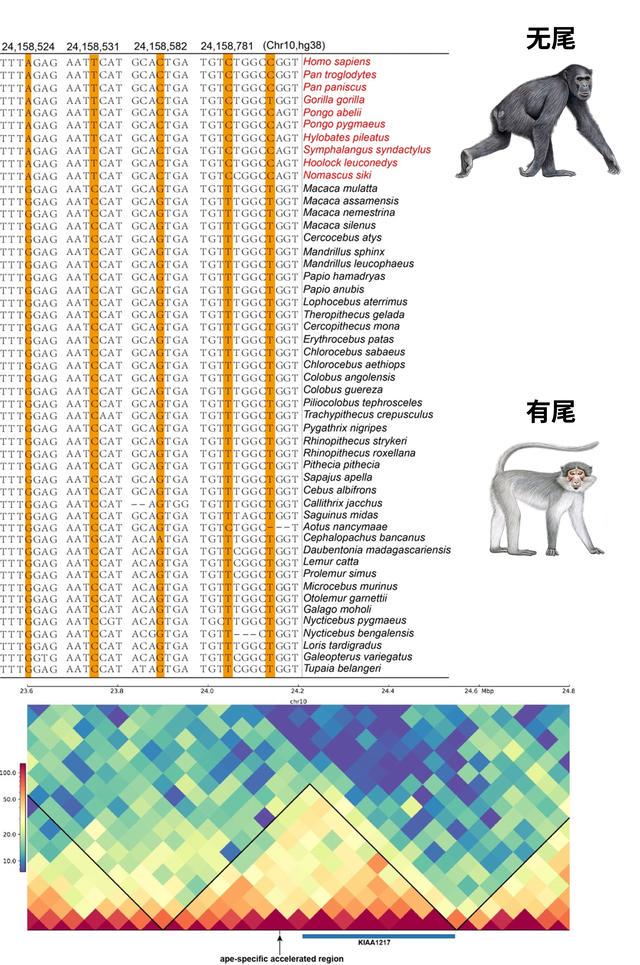
KIAA1217基因的调控区域在猿类中的快速演化可能导致其丢失尾巴。张国捷课题组和吴东东课题组合作供图
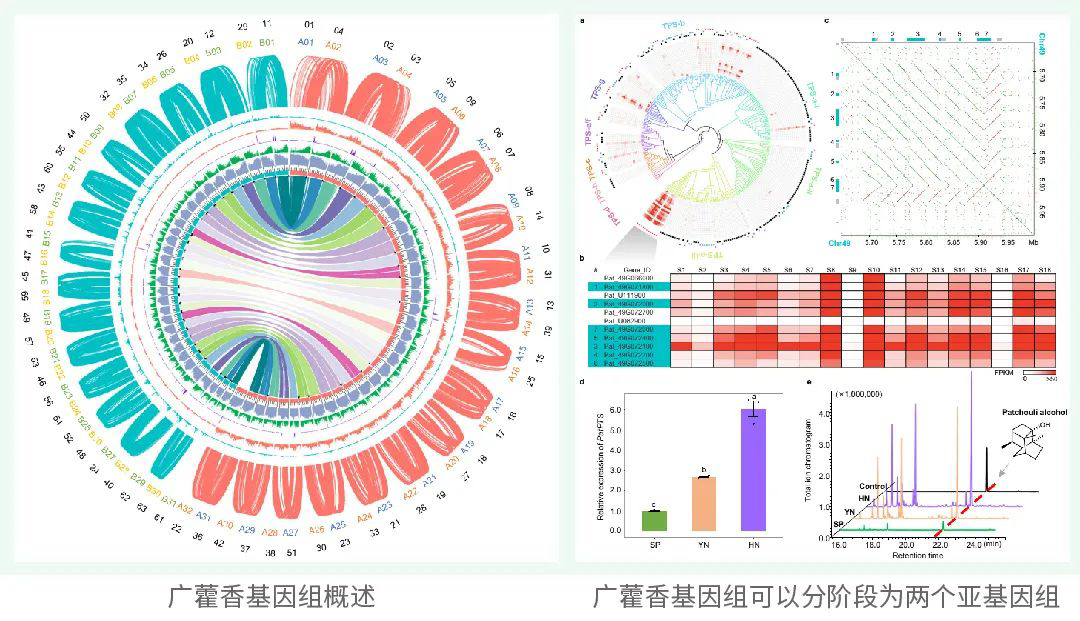
项目文章 | Cell!李家堂团队揭示蛇类的起源与演化机制
北京时间2023年6月19日晚,中国科学院成都生物研究所李家堂团队在《细胞》杂志上在线发表论文“Large-scale snake genome analyses provide insights into vertebrate development”。该论文基于大规模多组学技术与基因编辑等研究手段,全面揭示了蛇类起源及特有表型演化的遗传机制。
中国科学院成都生物研究所博士生彭长军、昆明动物研究所吴东东研究员和成都生物研究所助理研究员任金龙为该论文共同第一作者,成都生物研究所李家堂研究员为该论文的独立通讯作者。希望组为本研究提供了三代测序、NextDenovo(V 1.0)组装、NextPolish (V1.01)矫正和注释服务。


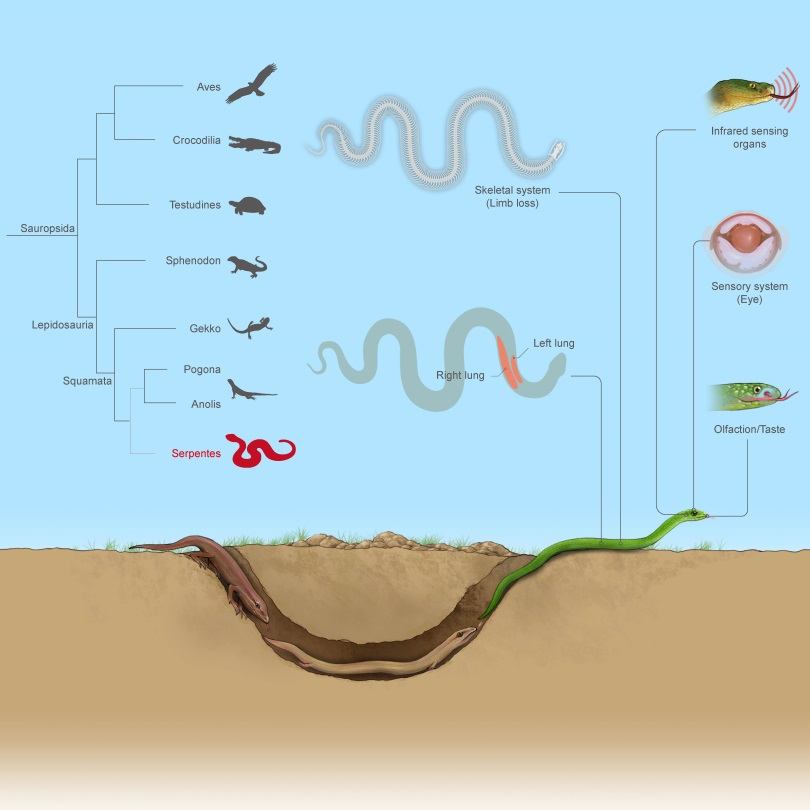
全球蛇类约4000种,广泛分布于除南极洲外的各大洲陆地和海洋,在进化历史上处于脊椎动物演化的关键节点,是脊椎动物的重要类群。蛇类演化出了四肢缺失、身体延长、左右肺不对称发育等特殊表型,揭示这些特殊表型的遗传机制对理解脊椎动物演化历史具有重要意义。
蛇类特有性状的演化遗传机制
基于染色体水平蛇类基因组数据集构建了迄今最有力的蛇类系统发育框架,推断蛇类起源于约1.18亿年前早白垩纪,支持了蛇类是由蜥蜴演化而来的假说。
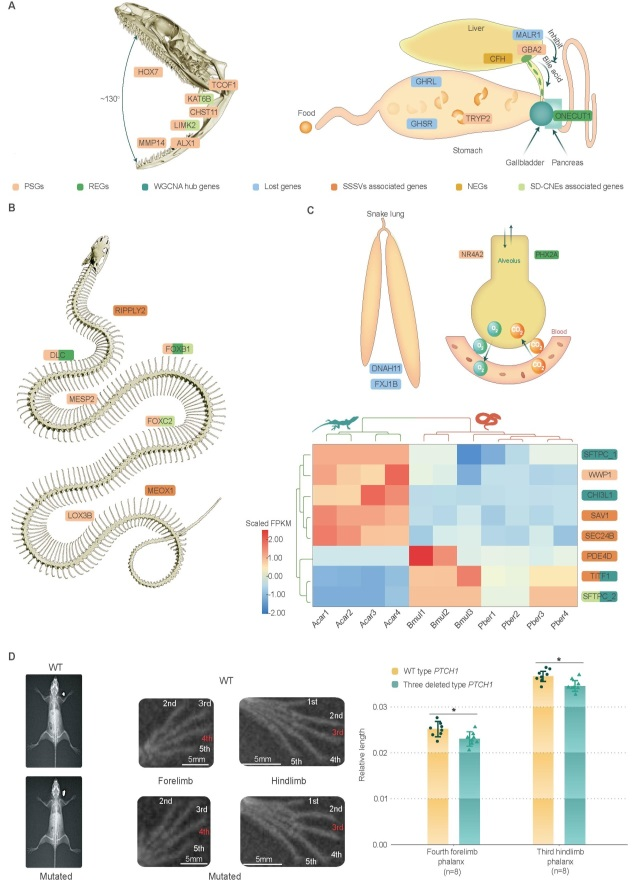
蛇类四肢缺失、身体延长、内脏器官不对称发育等重要遗传机制
基于谱系基因组与基因编辑等研究发现,蛇类PTCH1蛋白特异性缺失的三个氨基酸残基可能是其四肢缺失的重要遗传机制之一。大量编码及非编码调控元件的快速演化驱动了蛇类身体的延长。为适应身体延长,蛇类的内脏器官发生了不对称发育,如其左肺大多趋近于退化,而右肺则较为发达。蛇类丢失了控制器官对称发育的DNAH11和FXJ1B基因,是其左、右肺不对称发育的重要遗传因素。
此外,研究还探讨了红外感应蛇类和穴居的盲蛇类物种特殊表型的演化遗传机制。研究发现与热响应相关的PMP22基因和与三叉神经发育相关的NFIB基因的非编码调控元件的趋同演化是部分蛇类能够感知红外光谱的重要遗传驱动力。而盲蛇类物种则通过RPGRIP1等基因的丢失及CHIA等基因的快速演化以适应穴居生活,并形成专食蚂蚁及蚂蚁卵的食性。
李家堂团队未来将聚焦开发玉米蛇为模式动物并开展演化发育生物学研究。同时,围绕蛇毒等重要遗传资源的挖掘和运用,为抗蛇毒血清及蛇毒衍生药物的研发提供科学支撑。研究团队将推动从基础科学到应用基础科学的发展,更好地服务国家重大战略需求。
本研究得到中国科学院B类先导科技专项、国家自然科学基金等项目的资助。
相关论文信息:https://doi.org/10.1016/j.cell.2023.05.030

























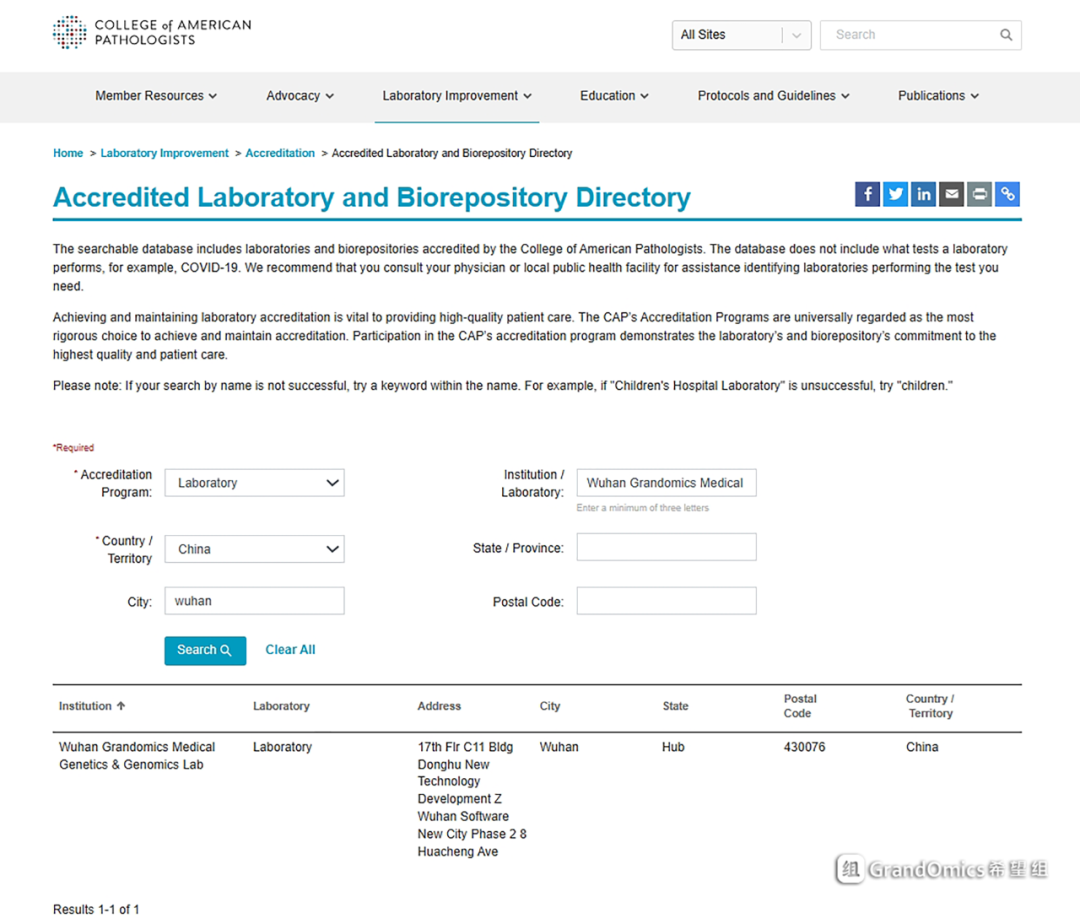 希望组医学实验室CAP证书
希望组医学实验室CAP证书

 CAP专家组现场评审及合影留念
CAP专家组现场评审及合影留念







