2022年9月22日,中国农业科学院作物科学研究所联合多家合作单位,在《自然遗传学(Nature Genetics)》杂志上发表了题为“Improved pea reference genome and pan-genome highlight genomic features and evolutionary characteristics”的研究论文。论文进行了豌豆参考基因组的组装和注释,进一步确定了全基因组变异,并基于全基因组重测序数据展示了 118 个栽培和野生豌豆基因型的种群遗传结构。通过基因组选择和数量性状位点(QTL)分析,发现了一批与驯化和育种改良性状相关的候选基因,其中包括孟德尔基因的几个候选基因。高质量的参考基因组和泛基因组为豌豆基因组进化和驯化提供了洞察力,并为豌豆遗传学和育种研究提供了宝贵的基因组资源。
中国农业科学院作物科学研究所杨涛副研究员和刘荣助理研究员、中国科学院微生物研究所骆迎峰副研究员和胡松年研究员以及山东省农业科学院农作物种质资源研究所王栋助理研究员为论文的共同第一作者。中国农业科学院作物科学研究所宗绪晓研究员、中国科学院微生物所高胜寒特别研究助理、山东省农业科学院农作物种质资源研究所丁汉凤研究员、国际半干旱热带作物研究所和澳大利亚默多克大学Rajeev K Varshney教授为论文的共同通讯作者。希望组为本研究提供了部分Bionano光学图谱服务。

豌豆 (Pisum sativum L., 2n=2x=14) 是一年生豆科植物,基因组大小约为 4.45 Gb。豌豆的收获面积在豆类中排名第四,仅次于大豆、普通菜豆和鹰嘴豆(http://www.fao.org/faostat/)。作为蛋白质、淀粉、纤维和矿物质的来源,由于其生物固氮能力具有显著的生态可持续性优势,豌豆一直受到关注,特别是自从孟德尔通过豌豆遗传试验揭示了遗传规律之后。豌豆被认为是最早驯化的豆科作物之一,然而,尽管它在推进植物遗传学方面发挥了关键作用,但其驯化过程仍然是一个谜,豌豆中栽培和野生豌豆的遗传多样性尚未完全揭示。
研究思路
部分研究结果
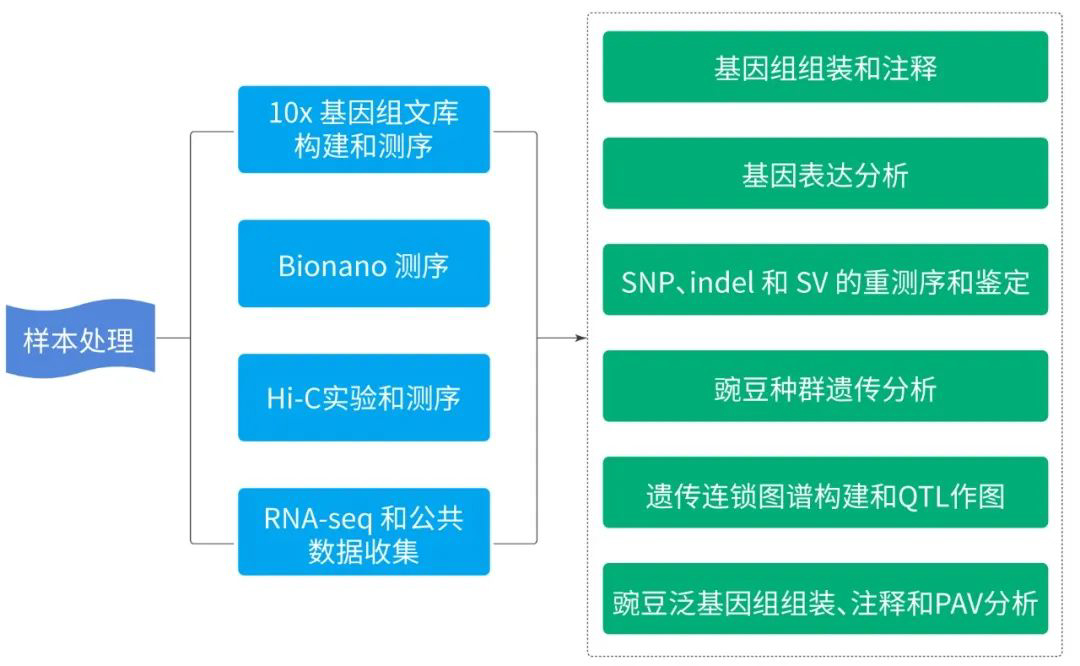
1.豌豆基因组图谱构建
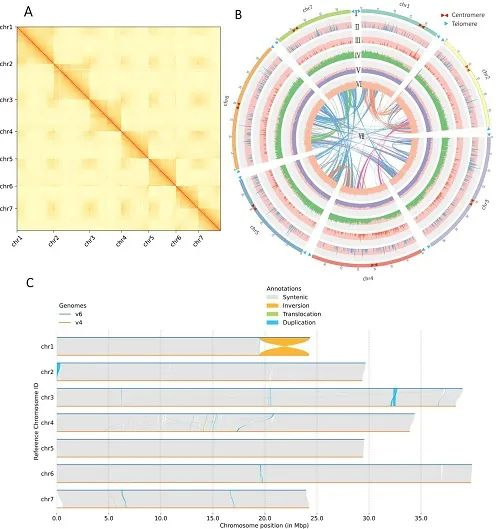
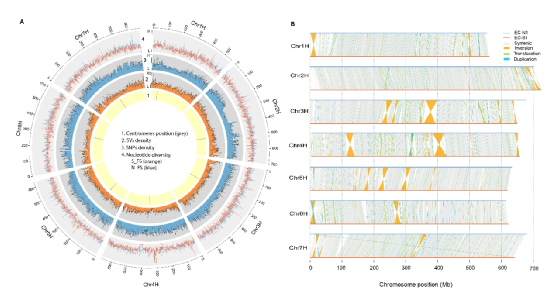
本研究结合使用 PacBio SMRT 测序、10x Genomics 、Bionano 光学作图、Hi-C 和 Illumina NGS 技术,对ZW6 的高质量、高连续性染色体参考基因组进行构建。最初基于 PacBio 读取的总大小为3,796.7Mb,contig N50 大小为 8.98Mb,最终组装被锚定到七个染色体水平的假分子中,具有两个细胞器基因组和 1,572 个未放置的重叠群(图 1 )。锚定重叠群的总大小为 3,719.6Mb,占豌豆ZW6 的 97.96%,而锚定重叠群仅占之前基于 NGS组装的 82.51%。豌豆基因组图谱的获得,为豌豆巨大基因组背后遗传学的了解奠定了基础。
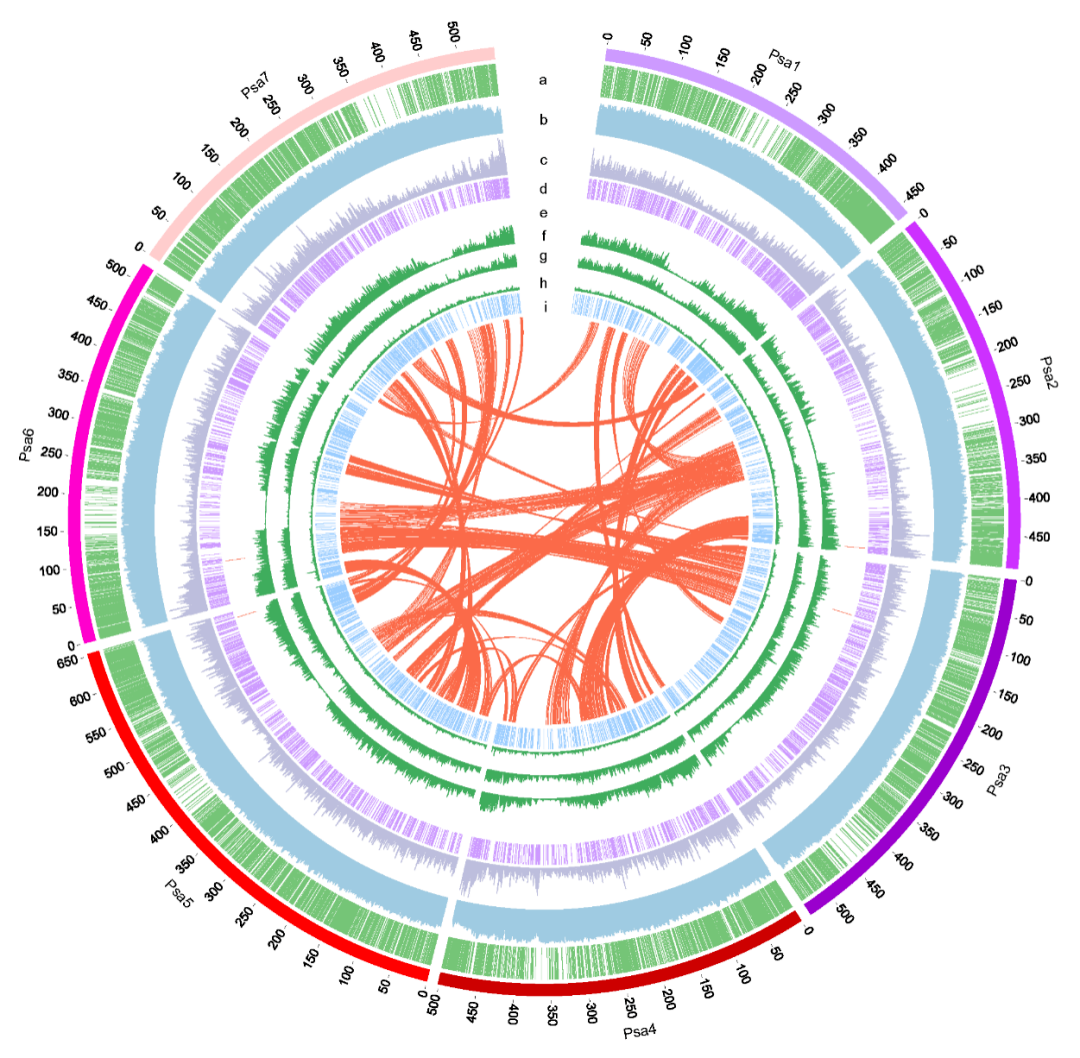
图1 豌豆基因组图谱
2.种群遗传结构
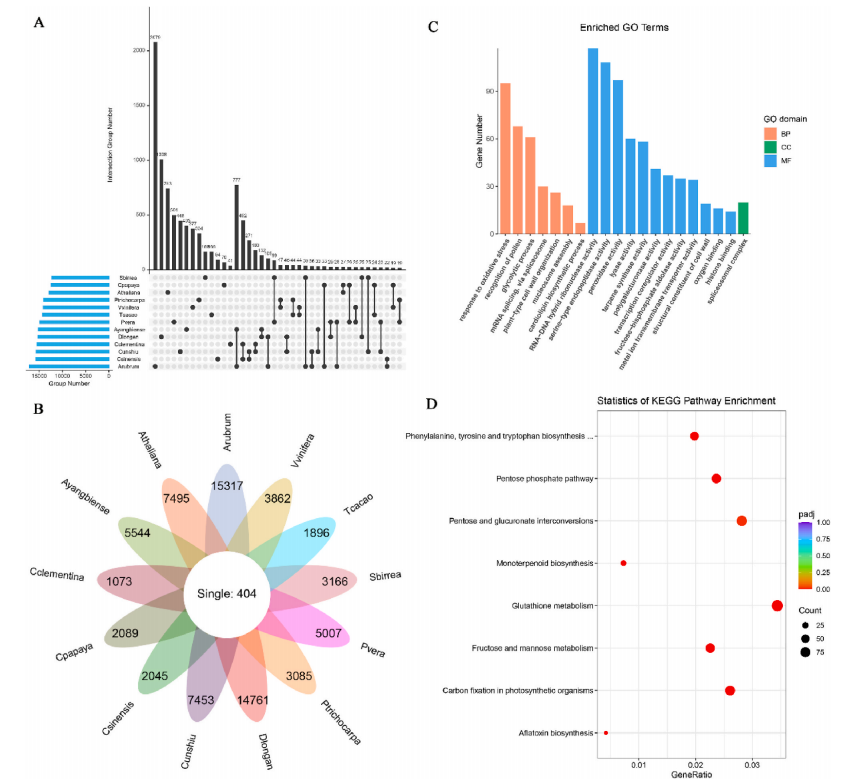
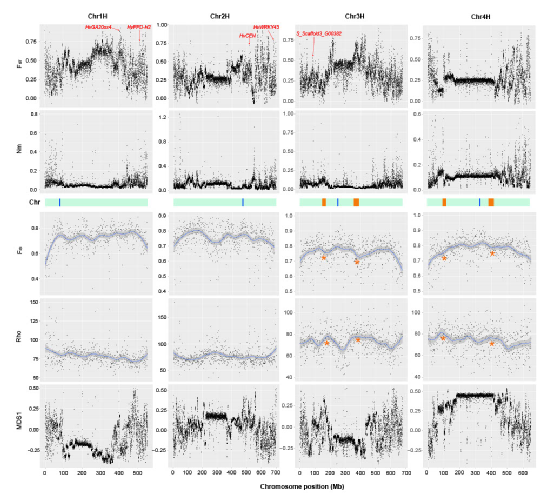
为了阐明豌豆中栽培和野生豌豆的系统发育关系和种群遗传结构,将 ADMIXTURE 应用于 SNP 和 SV 数据集,结果高度一致(图 2b、c )。P. fulvum、P. sativum 和 P. abyssinicum 三种不同种的结构得到了一致支持。在 P. sativum 中鉴定了三个遗传组,其中 P. sativum IV (PSIV)代表早期分化组(图2b,c)。P. sativum II (PSII) 和P. sativum III (PSIII) 主要对应于代表不同地理区域(即亚洲和欧洲)栽培豌豆的两个遗传组,这可能与豌豆驯化后的传播途径有关(图2b,c)。用 SNP 和 SV 数据集构建的系统发育树(图 2a,d)显示出主要分支的相似系统发育关系,并且与 ADMIXTURE 结果的主要遗传组有良好的对应关系。此外,P. fulvum、P. abyssinicum和栽培的 P. sativum的 Pisum 形成了三个独立的单进化枝(图 2a,d),这也得到了 SNP 和 SV 数据集的主成分分析的支持(图 2e, f )。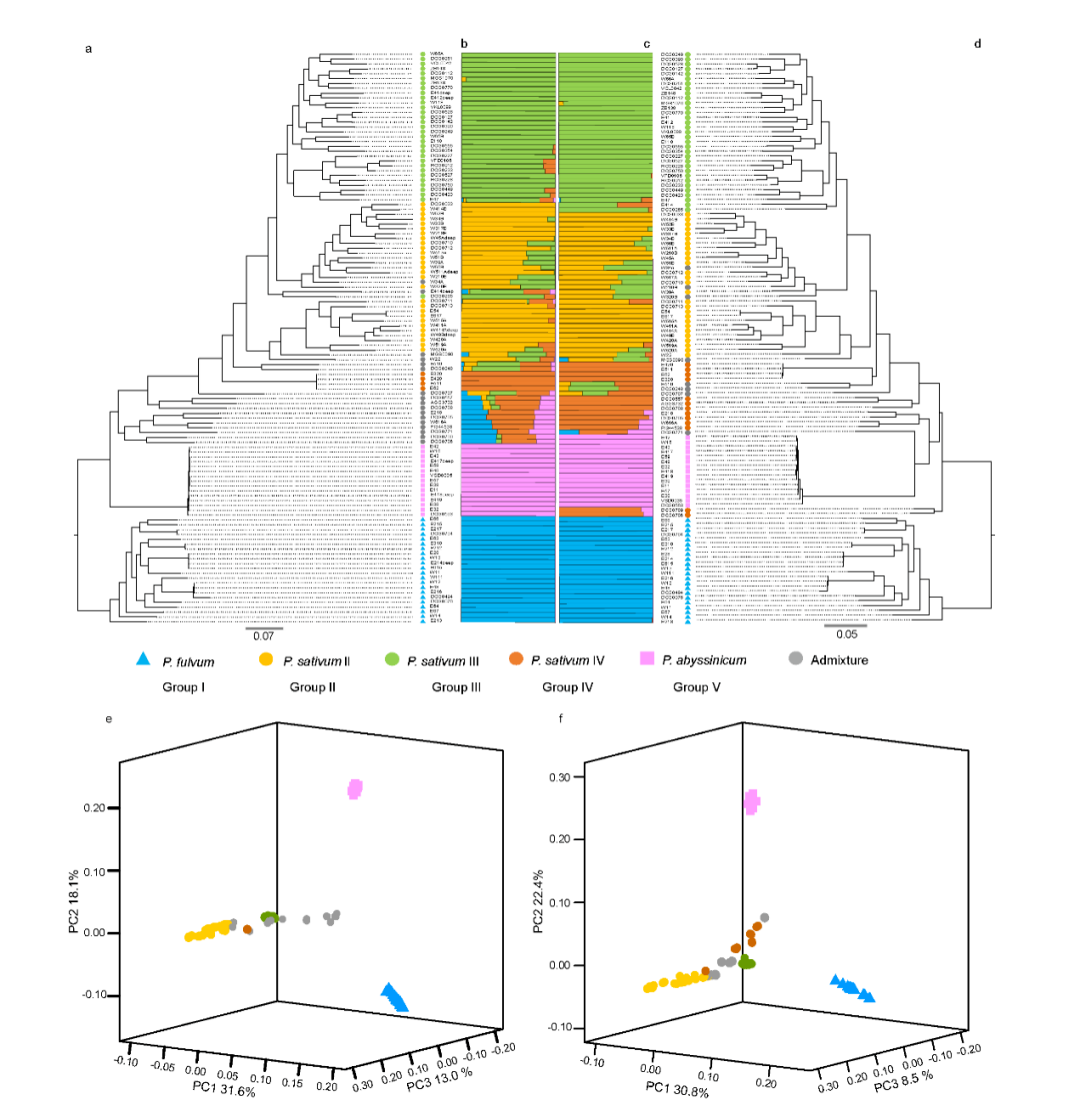
图2 基于SNP (a, b, e)和SV (c, d, f)的118份栽培和野生豌豆的群体遗传结构
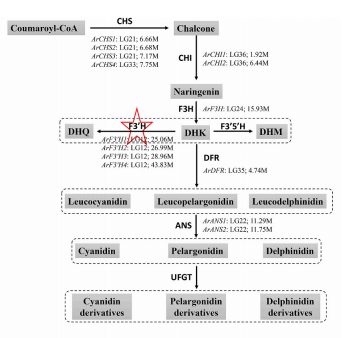
3.孟德尔基因位点的 QTL 分析和重新发现为了探索豌豆重要农艺性状的遗传基础,使用基因分型测序对 300 个 F2 种群(WJ×ZW6)中的 12 个农艺性状进行 QTL 分析。将总共 124,900 个高质量 SNP 标记聚集成 2,950 个 bin 标记,构建了一个高密度(0.31 cM)遗传连锁图谱,组装成跨越 924.1 cM 的七个连锁群。发现 25 个 QTL 与 12 种农艺性状相关,比值对数 (LOD) 值范围为 4.2 至 78.1,解释的最大表型变异 (PVE) 高达 68.7%(图 3a)。在 25 个 QTL 中,与 Mendel 分析的三个性状相关的 SS3、SL5 和 PF5 显示出更高的 LOD(78.1、53.1 和 31.9)和 PVE(68.7%、46.7% 和 37.6%),在基因组中具有尖锐的 QTL 峰(4.87Mb, 1.85Mb 和 4.43Mb)(图 3b-d)。SS3、SL5 和 PF5 中的同源比对和功能注释的结果发现了两个先前已知构成孟德尔性状对应的基因位点,R和 Le,以及一个可能与荚型相关的候选基因。然而,这些基因都没有落在推定的选定区域中,这意味着它们可能与豌豆驯化没有密切关系(图3e-g)。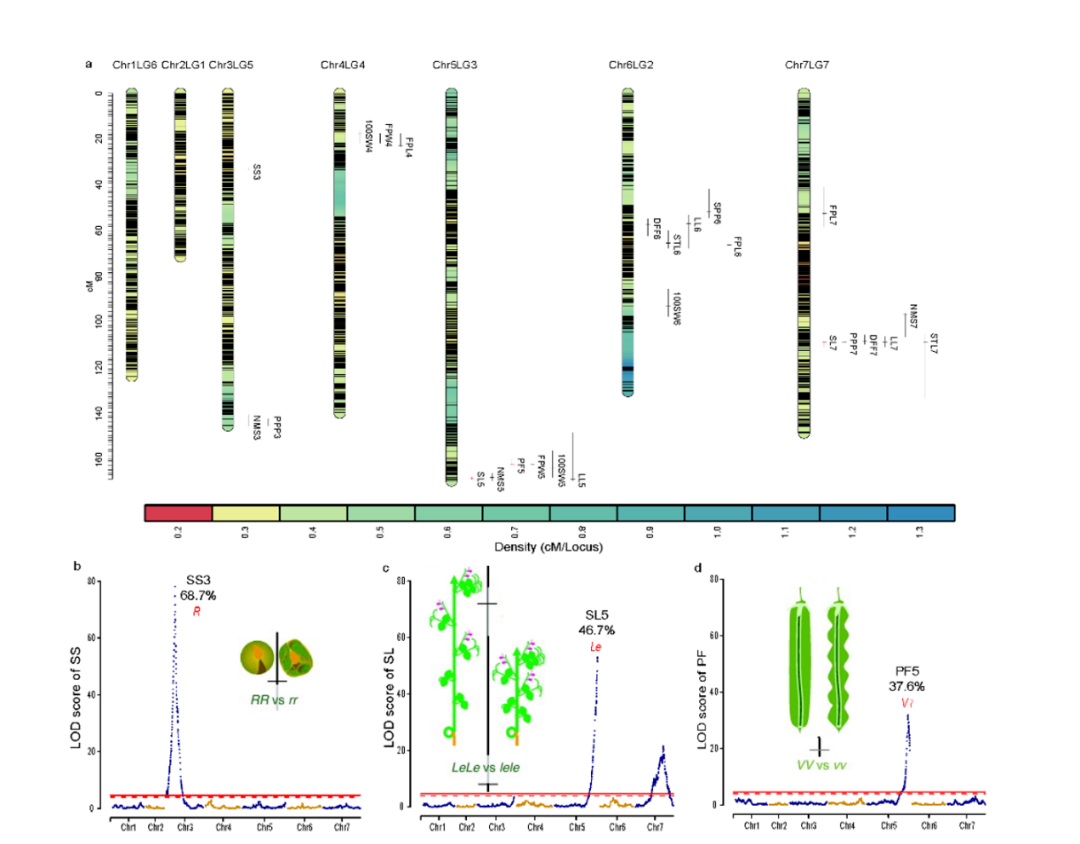
图3 基于SNP (a, b, e)和SV (c, d, f)的118份栽培和野生豌豆的群体遗传结构
4.基于 118 个栽培和野生豌豆的泛基因组
随着新基因组的增加,核心基因的数量减少,而泛基因的数量增加,逐渐趋于饱和(图4a)。在质量控制之后,基于跨基因组直系同源物的系统发育,116个基因组的基因被聚集成 112,776 个泛基因,代表系统发育分级直系群(HOG)(图 4)。Pisum中核心基因、软核基因、壳基因和云基因的数量分别为15,470、6,170、41,028和50,108,分别占预聚类基因总数的35.19%、15.54%、44.28%和4.99%。任何组中核心基因的百分比均高于 Pisum 整体。值得注意的是,群体的核心百分比可能与其计算的遗传多样性相对应,这表明遗传多样性也可能对核心基因的百分比有贡献。同时,核心基因在其他 27 个植物基因组中也更保守(图 4b ),表明它们在基本功能中的作用。此外,PAV 的邻接树也显示出 116 个 Pisum 种质的明显分离,这与基于 SNP 和 SV 的结果高度一致,表明有助于 Pisum 驯化的重要遗传变异也存在 PAV 中。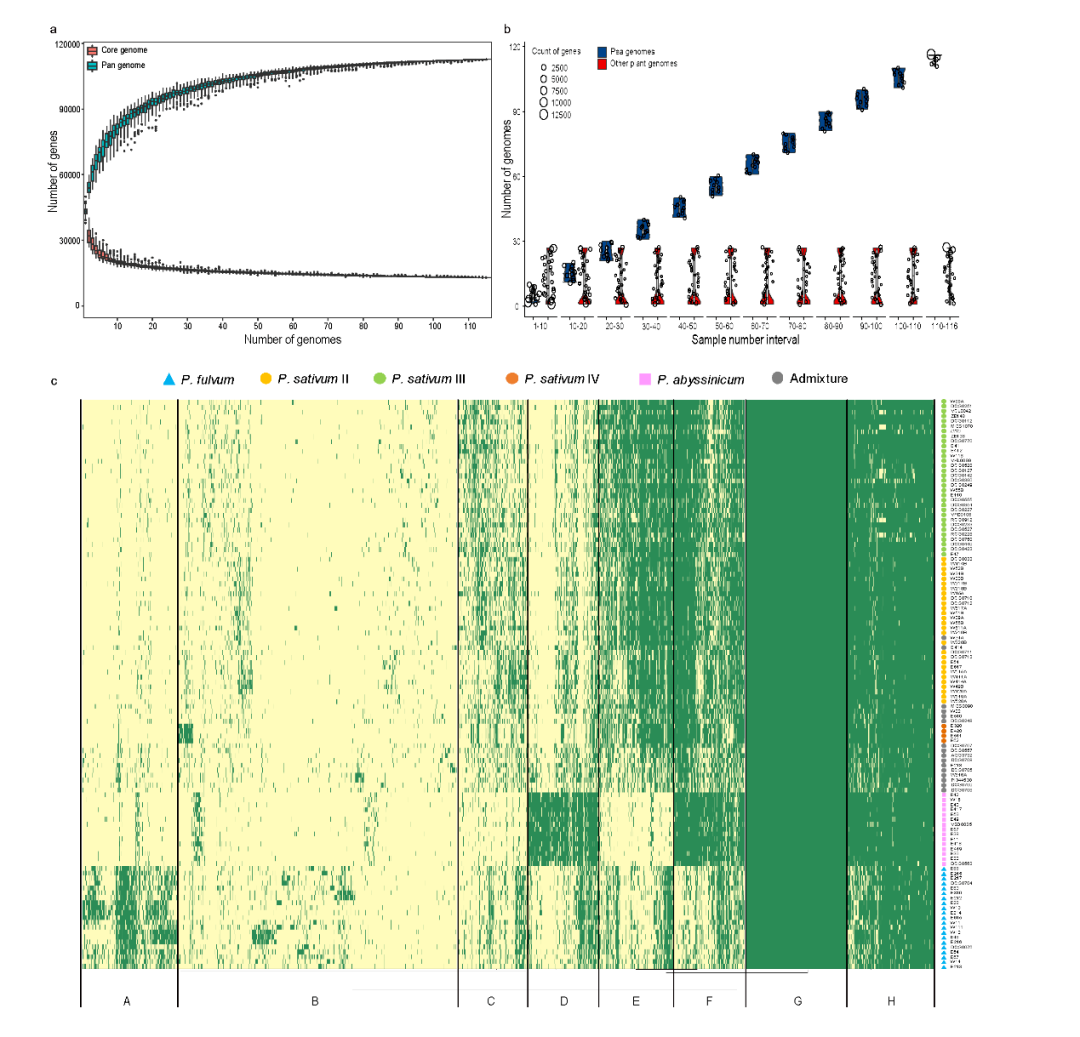
图4 116个代表性栽培和野生豌豆的泛基因组分析结果
总之,这里介绍的高质量参考基因组和泛基因组提供了对豌豆基因组进化和驯化的见解,以及豌豆遗传学和育种研究的宝贵基因组资源。这项研究将填补以前的遗传模式生物和现代基因组学之间的空白,以促进豌豆的研究和作物改良。








 注:在3个case中OGM未检出位于X染色体PAR区域的P2RY8::CRLF2融合,可能源于其VAF小于5%的检测下限,预计可在后续算法软件提升中解决该问题。
注:在3个case中OGM未检出位于X染色体PAR区域的P2RY8::CRLF2融合,可能源于其VAF小于5%的检测下限,预计可在后续算法软件提升中解决该问题。