种子植物包括裸子植物(gymnosperms)和被子植物(angiosperms),裸子植物分为四大类,即苏铁类(cycads)、银杏类(Ginkgo)、松柏类(conifers)和买麻藤类(gnetophytes)。裸子植物基因组较大,重复序列含量高,结构复杂,迄今为止,现存最原始种子植物苏铁分支尚缺少完整的基因组图谱。

2022年4月18日,由22个机构65位科学家联合在Nature Plants发表了题为“The Cycas genome and the early evolution of seed plants”的封面文章,报道了现存最原始种子植物苏铁参考基因组,填补了种子植物基因组研究的空白。苏铁基因组的发布,代表着种子植物基因组演化研究中的最后一块拼图完成,为后续比较基因组学的开展奠定了基础。希望组参与了本研究项目中攀枝花苏铁的测序、组装及初步注释服务。

苏铁基因组测序材料选取苏铁类的基部类群、也是整个苏铁类分布纬度最北的种类攀枝花苏铁(Cycas panzhihuaensis)。基于长片段测序与MGI-SEQ测序,苏铁基因组组装大小为10.5 Gb,contig N50为12Mb,结合Hi-C数据,挂载为11条染色体。其中共注释32,353个蛋白编码基因,BUSCO评估完整度为91.6%,是目前裸子植物中最高质量的大基因组图谱。
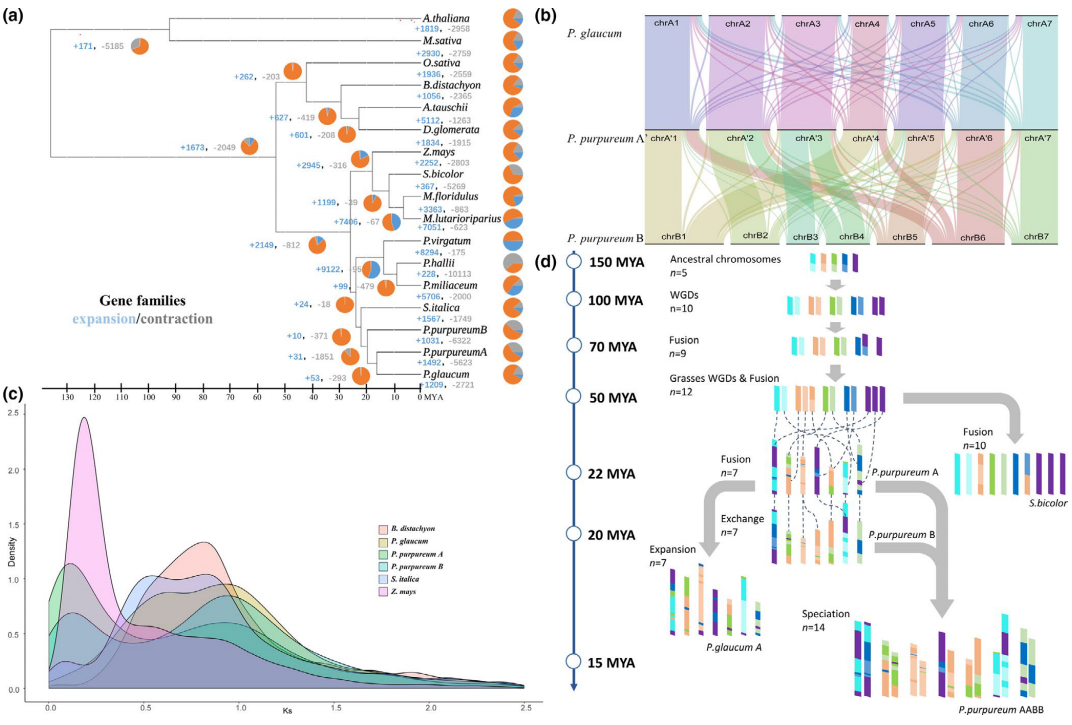
裸子植物具有4大分支,1118种,关于裸子植物内部大分支之间的系统演化关系一直有不同的学术观点。基于15个维管植物基因组3282个直系同源低拷贝核基因、90个种子植物转录组1569个直系同源基因、72种维管植物叶绿体和线粒体基因组数据的系统分析结果表明,苏铁单独(线粒体数据)、或和银杏一起(核基因、叶绿体数据)构成其它所有裸子植物的姐妹群。
基因组加倍是植物演化适应的重要驱动力,关于裸子植物共同祖先是否经历了全基因组加倍事件一直存在争议。研究者采用对重复基因同义替代分析和系统发育基因组学方法,并使用基因组内共线性区域进行比较验证,发现现存裸子植物的最近共同祖先可能经历了一次古老的全基因组复制事件(命名为ω,图一a)。伴随着种子植物起源,许多关键创新性状如种子发育、花粉、次生生长相关的基因家族均发生了创新或扩张。在种子植物的祖先节点共发现663个新获得的基因家族和368个扩张的基因家族。其中,106个新获得和55个显著扩张的基因家族与种子生理发育有关,包括调控胚胎早期发育、种子休眠和萌发、种子能量和营养代谢,种皮形成以及种子的免疫和应激反应等(图一b)。
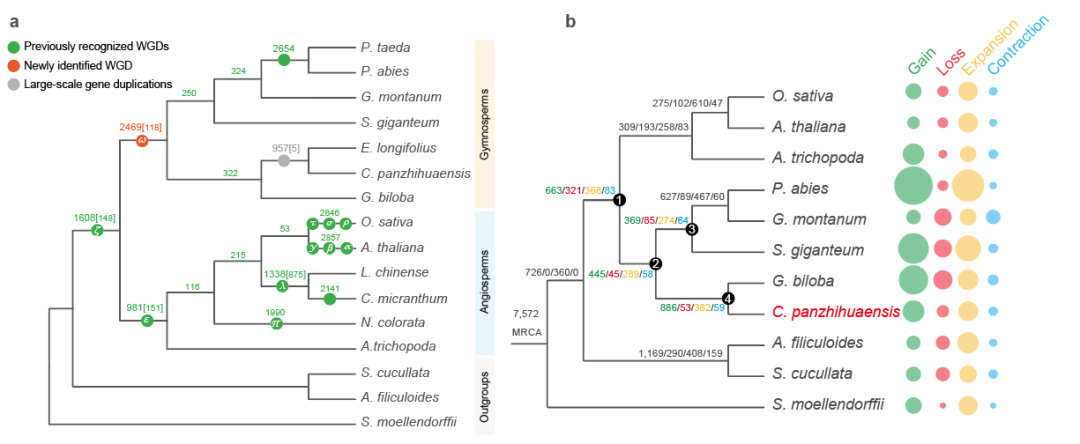
图一、a) 基于系统发育关系推断种子植物的全基因组加倍事件;b) 种子植物的基因家族创新和扩张。
最显著扩张的种子生理相关家族是cupin蛋白家族。攀枝花苏铁编码一类新的vicilin-like贮藏蛋白 vicilin-like antimicrobial peptides(v-AMP),在基因组中呈串联基因阵列分布,多在授粉胚珠后期和受精胚珠时期表达,而后逐渐降低,暗示v-AMP基因在种子发育过程特定时期发挥重要作用。LAFL家族(LEC1、ABI3、LEC2和FUS3)是种子发育核心调控基因,苏铁等裸子植物的FUS3和LEC2基因可构成一个新的进化枝,定义为FUS3 / LEC2-like类型,与被子植物的FUS3和LEC2形成姐妹分支关系。FUS3 / LEC2-like类别是裸子植物特有的。在攀枝花苏铁授粉后,其会表现出明显的表达,表明可能在裸子植物胚胎发生早期发挥特定作用 。
苏铁类起源于古生代二叠纪早期,距今已有至少2亿7千万年历史。在经历大量灭绝以后,现代苏铁多是近期几次辐射演化的后代。如今苏铁具有2科10属。研究者基于现存苏铁目339种植物的转录组数据,重建了苏铁类自身的系统发育关系。分子钟分析表明,现存苏铁的多样化同步发生于距今1100至2000万年之间,是中新世以来气候剧烈变化的结果(图二)。
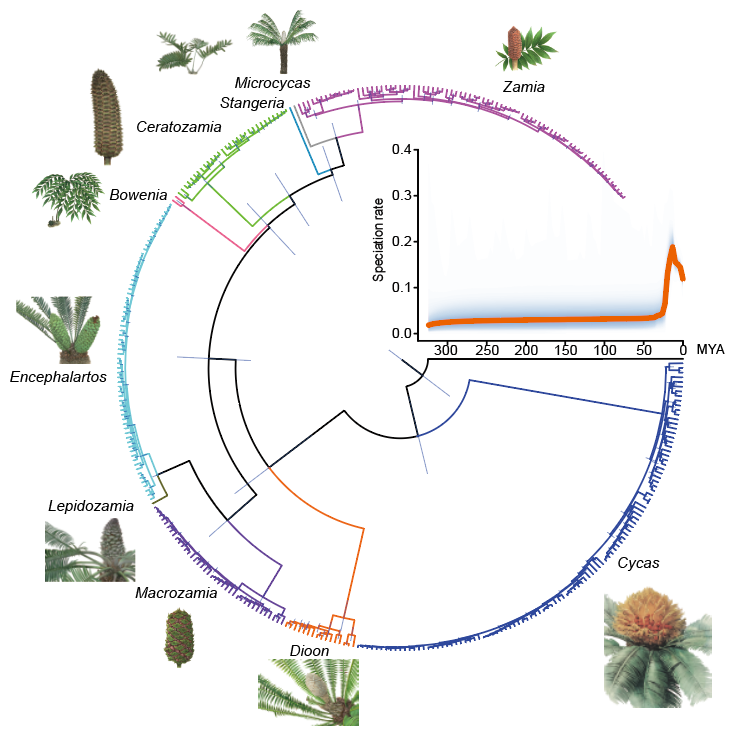
图二、苏铁目系统发育树支持现存苏铁是辐射演化的结果
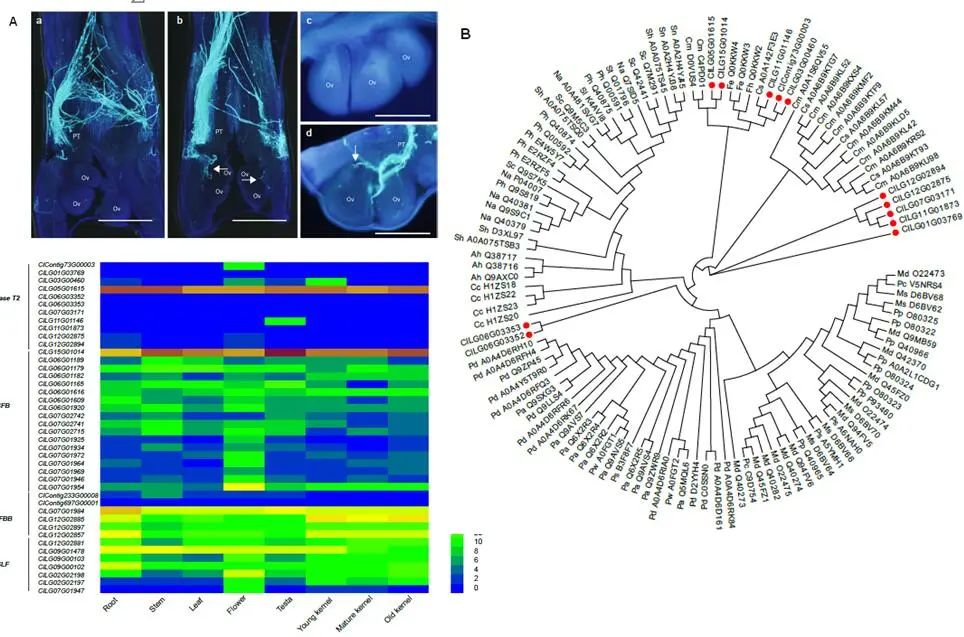
雌雄性别分化是一种进化性状。裸子植物已报道的1118个物种中,65%的种类都是雌雄异株, 而在被子植物中雌雄异株只占到6%。苏铁类植物均为雌雄异株(图三),由于其生长缓慢,以往只能在开花时才能判断性别,而成株树龄多在十年以上以及在适宜的环境才会开花。研究苏铁性别决定的分子机制,可以在植株进入花期前就确定性别,对于苏铁类植物的就地和迁地保护和园林培育具有重要意义。苏铁的性别控制基因一直困扰科学界。研究者通过对源于四川攀枝花苏铁国家级保护区62株雌雄苏铁群体测序,表达差异分析,和雄性Y染色体的组装,找到雌雄表达差异最大的一个基因来自雄株的Y染色体,该基因编码一个MADS-box转录因子,推测其调控雌雄苏铁的性器官发育,揭示了苏铁性别决定的遗传机制。该转录因子的同源基因也仅能在雄株基因组中检测到,说明了该性别决定机制在苏铁类植物中的保守性。
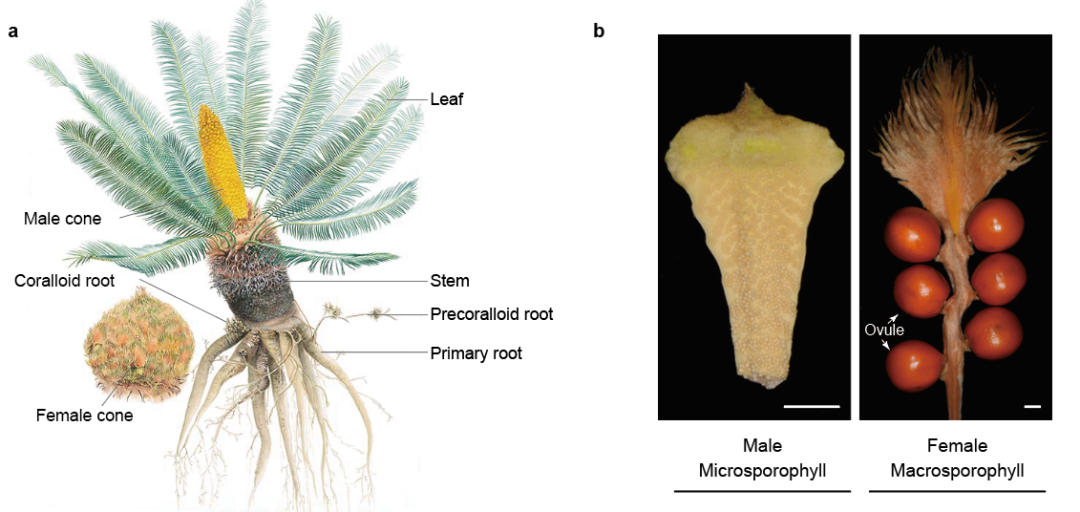
图三、a) 攀枝花苏铁;b) 攀枝花苏铁雄株和雌株的孢子体。
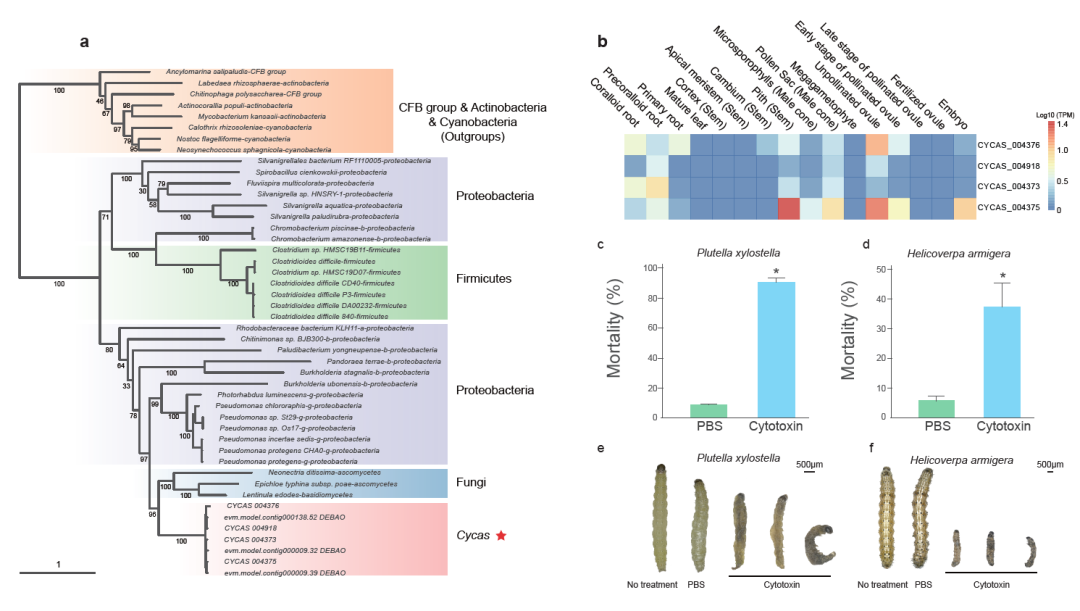
早期维管植物的精子都是有鞭毛,可以游动的。随着演化,鞭毛丢失。在现生种子植物中仅苏铁和银杏保留精子具鞭毛的特征。研究人员发现,苏铁和银杏均保留了大量鞭毛组装所需基因,但与苏铁相比,银杏中RSP类基因有一定的丢失(RSP2, RSP3, RSP 9, 和RSP11等)。此外,与鞭毛行使功能密切相关的外周致密纤维合成基因(ODFs),只在苏铁和银杏基因组中存在,在其它种子植物中则全部丢失。进一步证实了苏铁在种子植物演化中古老的地位。水平基因转移泛指不同物种间的基因交流,在陆生植物适应性进化过程中起到推动作用。研究者在攀枝花苏铁基因组中发现一种细胞毒素蛋白基因(fitD),这种基因起源于细菌,通过水平基因转移的方式转移到真菌和苏铁中(图四a)。基于苏铁类339个物种转录组数据,研究人员发现该毒素蛋白只在苏铁属物种中存在。同时,fitD基因在种子和根部高表达,这可能也是苏铁种子和根部具有毒性的原因之一。基于基因重组技术,在大肠杆菌表达的毒素蛋白产物对小菜蛾和棉铃虫有显著致死性(图四b-f),显示出毒素蛋白具有一定的农业应用前景。 图四、a)苏铁水平转移毒蛋白基因的演化历史。b-f) 苏铁毒蛋白基因表达,及对昆虫毒性实验。
该研究由深圳华大生命科学研究院、深圳市仙湖植物园、中国科学院昆明植物研究所、兰州大学、中国环境科学研究院、河南大学和南京林业大学等22个机构65位科学家联合完成。该论文第一作者为深圳华大生命科学研究院刘阳、王思博、李林洲、杨婷、魏桐,深圳仙湖植物园董珊珊,兰州大学武生聃等为共同第一作者,分别在基因组不同的领域贡献了自己的专业力量。深圳仙湖植物园张寿洲,深圳华大生命科学研究院刘欢,中科院昆明植物所龚洵,美国佛罗里达大学Douglas E. Soltis,比利时根特大学Yves Van de Peer为文章共同通讯作者。该项目得到深圳市城市管理和执法局科研专项、国家重点研发计划、生态环境部生物多样性调查与评估等基金支持。





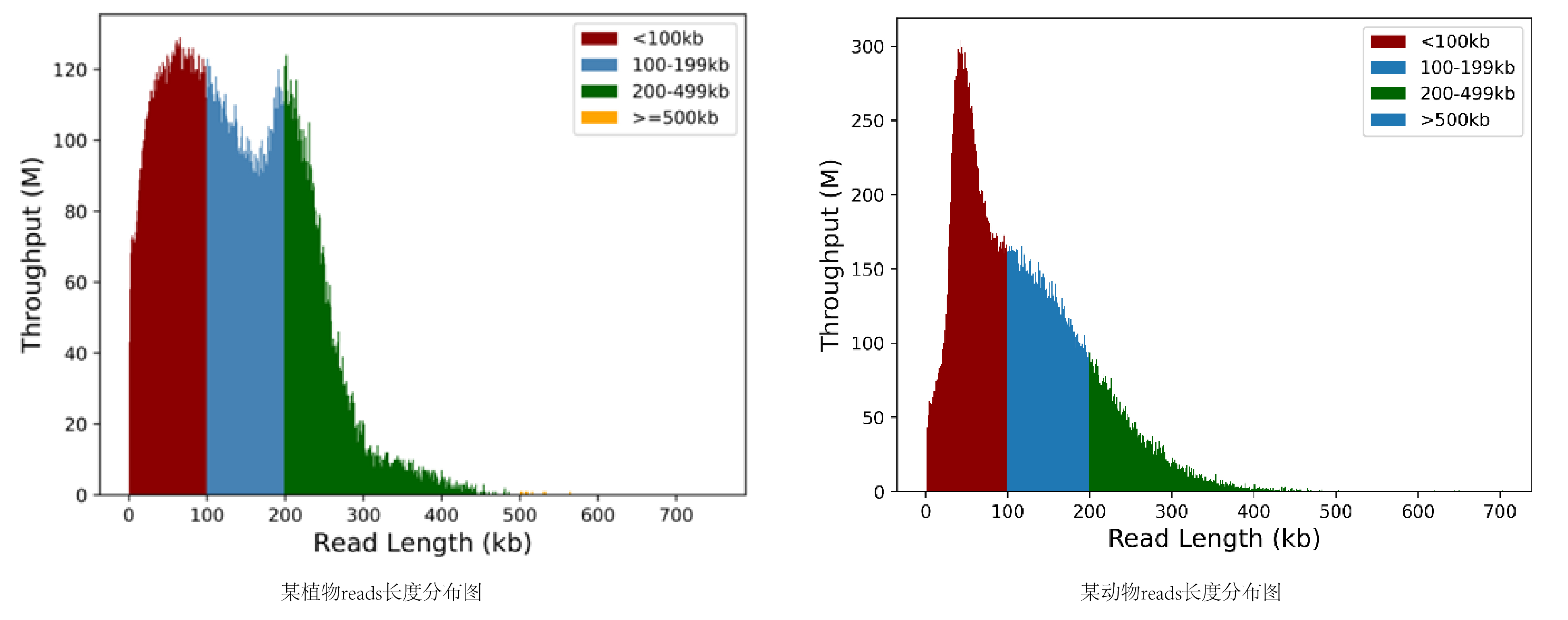
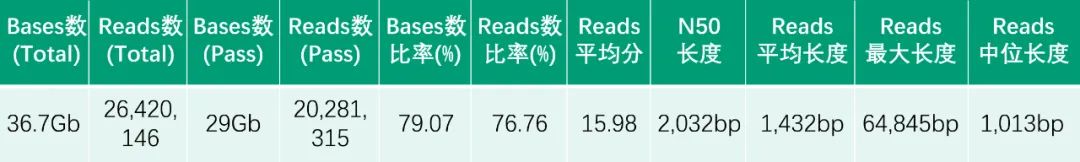
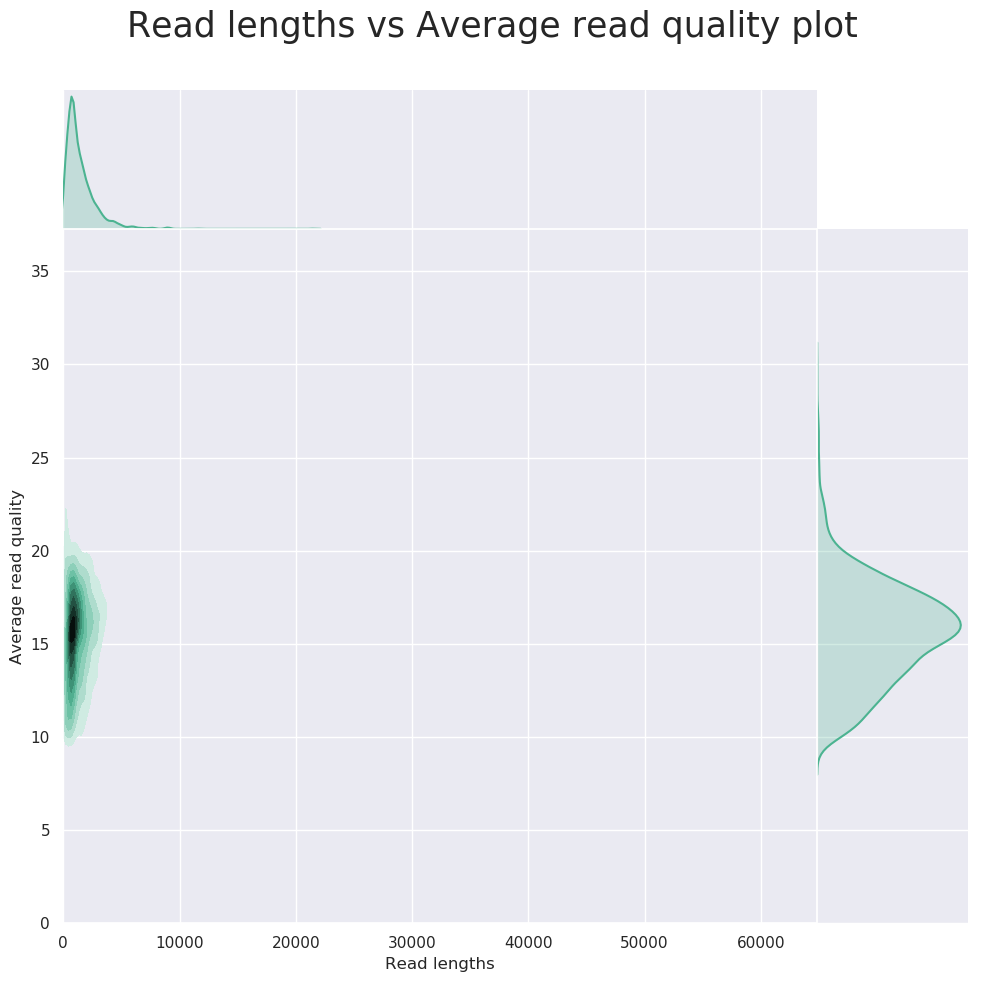
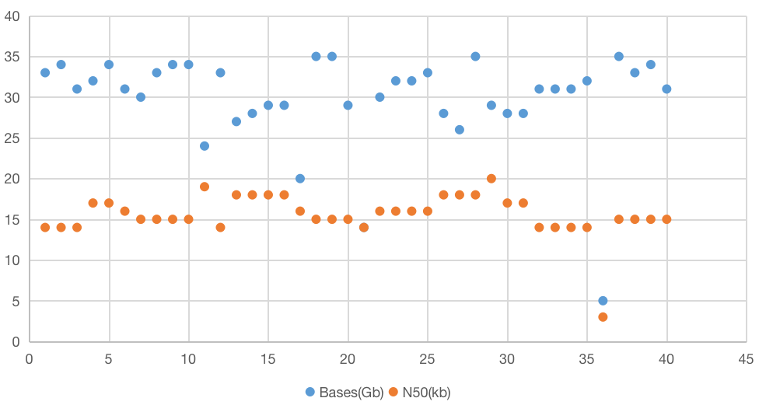 HiFi数据量和长度统计
HiFi数据量和长度统计


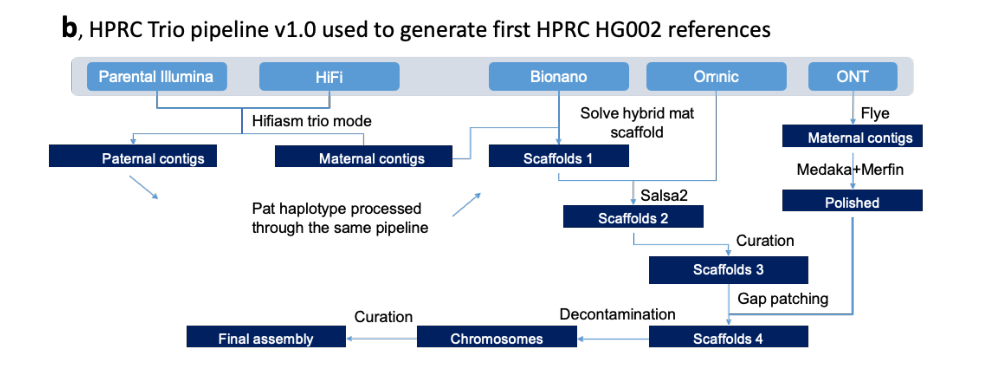 HPRC Trio pipeline v1.0组装流程图
HPRC Trio pipeline v1.0组装流程图