武汉希望组被认定为湖北省专精特新“小巨人”企业


近日,北京希望组与Sentieon联合宣布,双方决定在基因组数据分析的多个领域进行战略合作,持续推动包括二代和三代测序在临床诊断的产品落地。
双方合作的重点包括了以下四点:
3.验证并部署基于Sentieon已经完成的MGI数据全基因组(WGS)分析流程;
4.将双方的合作纳入“希望诊断计划”,为该计划开发并搭建全方位的长、短读长结合的大数据变异分析平台。
高质量的结构变异检测,是基因组医学的基石,无论是针对罕见病、肿瘤,还是辅助生殖,各种基因组医学的应用,均急切的需要新型的工具解决复杂的基因组结构变异检测问题。
长读长测序对于大片段的结构变异检测具有天然的优势。综合长读长数据分析算法中的序列拼接和序列比对算法,希望组开发了针对于长读长测序数据(PacBio HiFi和ONT)的SV检测软件——GrandSV。和以往长读长数据SV检测软件相比,兼顾了两种特征鲜明、截然不同的长读长数据(PacBio HiFi/ONT),具有更高的准确度和灵敏度。
利用准确度在99%左右的PacBio HiFi模拟数据来评估GrandSV与同类型软件cuteSV v1.0.9,Pbsv v2.4.0,Sniffles v1.0.12在人类基因组上鉴定结构变异(SV)以及变异分型(Genotyping)的准确性和灵敏度。图1的结果显示,GrandSV在5-30X的不同深度上效果都是最好的之一。此外,模拟Super Accuracy basecalling模式下的ONT数据分析结果也同样显示出了GrandSV的不俗表现。
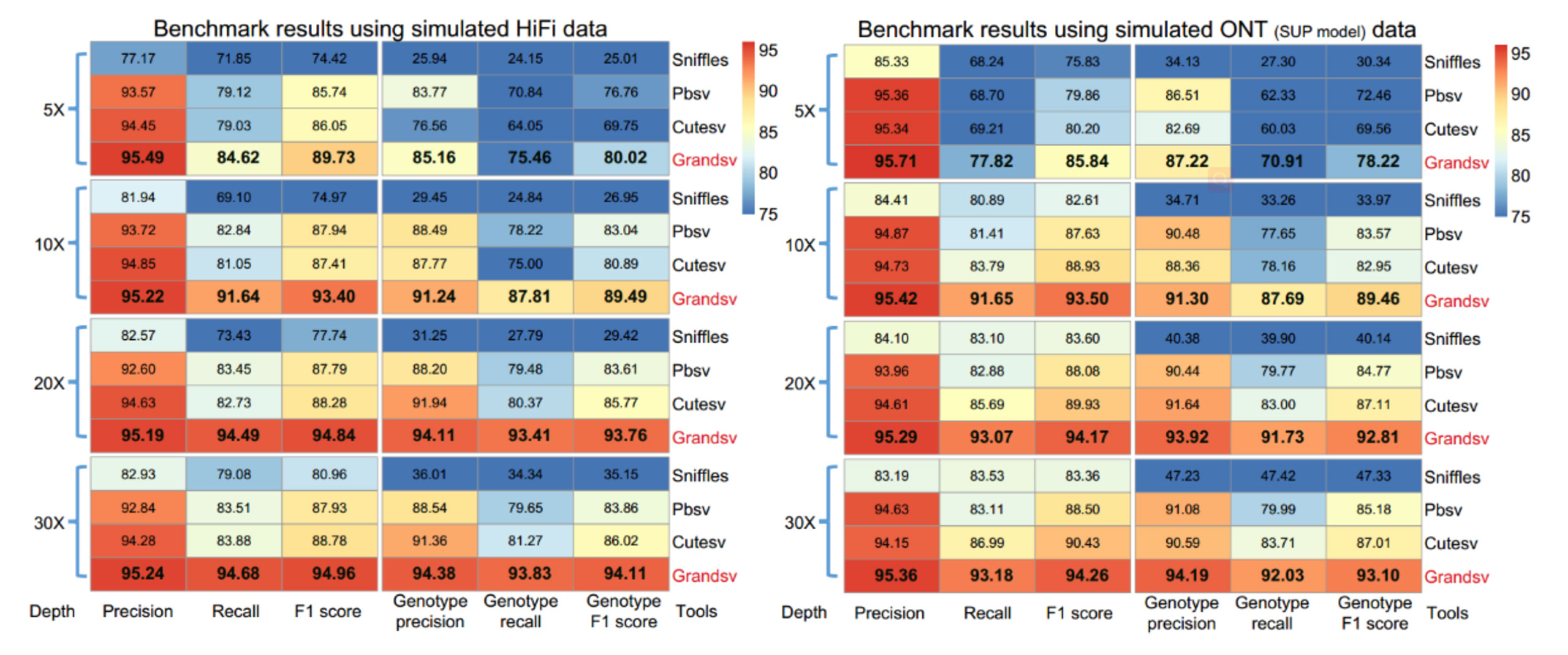
图1. Benchmark with simulated PacBio HiFi and ONT (SUP model) data
除模拟数据外,基于HG002的PacBio HiFi和ONT真实数据,并以Genome in a Bottle Consortium (GIAB) 团队发表的34,830个高置信区间当做真实的背景数据集来评估GrandSV表现。结果依然显示GrandSV的整体效果在5-30X的不同深度上都是最优的之一。
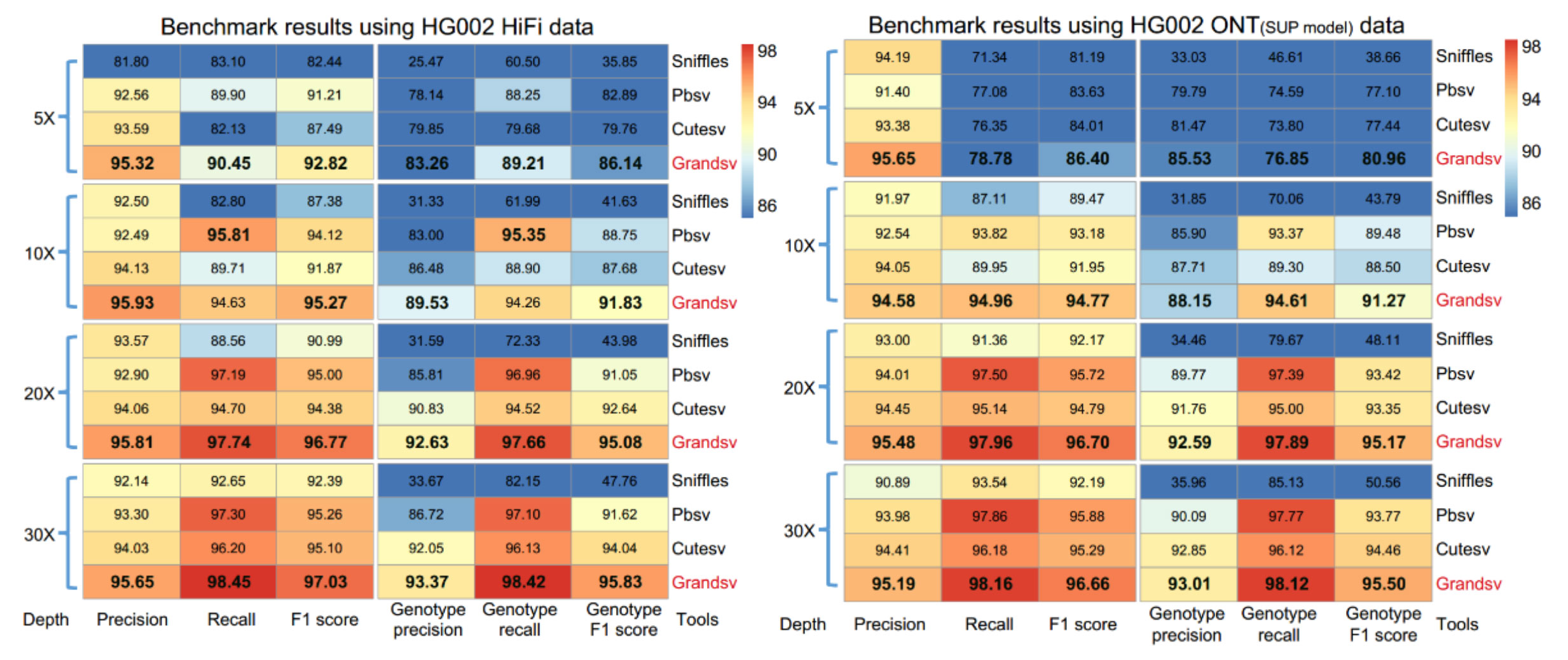
图2. Benchmark with HG002 PacBio HiFi and ONT data
相较于其他长读长SV检测软件,GrandSV有着更高的灵敏性。图3是HG002中一个长的片段插入,由于测序reads太短,单条read无法跨过,导致其他的软件在此处只能检出断点,而GrandSV通过局部组装可以完整的组装出跨过这个SV的一致性序列。图4是HG002中两个杂合的片段插入,其他软件只能检测出一个平均长度约94bp的片段插入,而GrandSV能够准确检测出两个不同的杂合的片段插入。
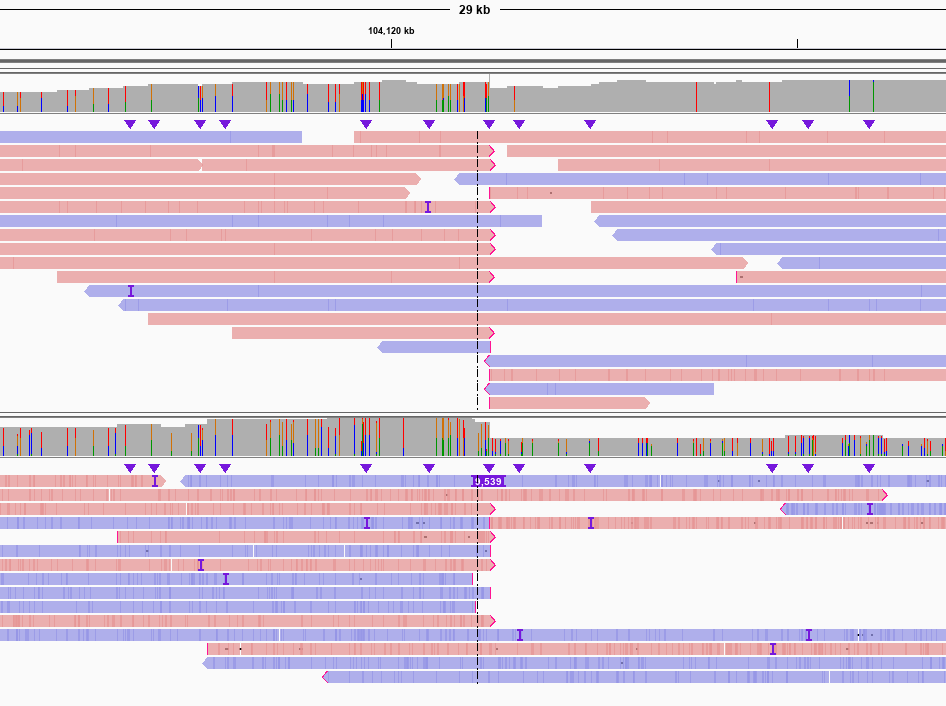
图3. Only GrandSV called correctly for a 9562 bp INS
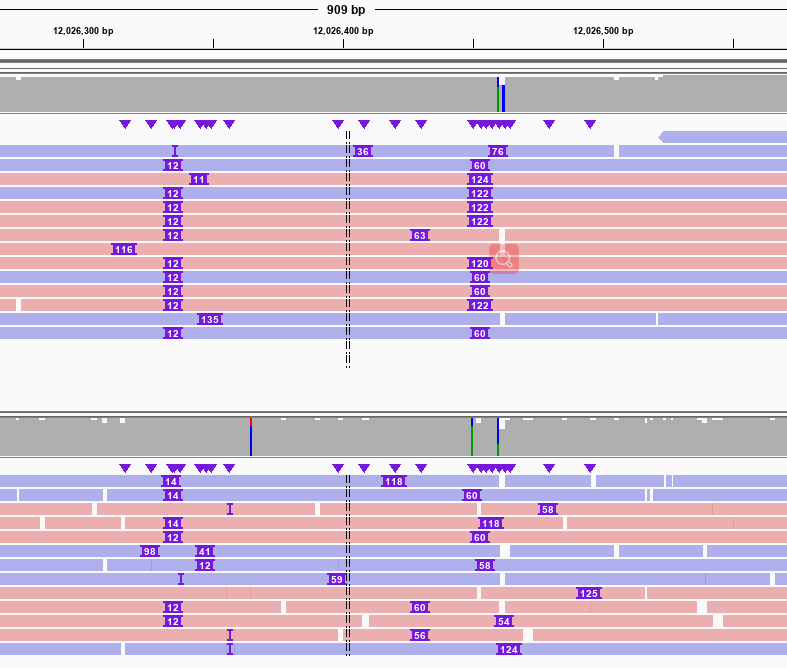
图4. Only GrandSV called correctly for a 60 bp INS and a 122 bp INS
Sentieon在二代测序中SNP/Indel变异检测流程已非常成熟,并以其检测准确性高和检测速度快而广受业内人士认可。近日,Sentieon推出了DNAscope LongReads分析流程,深度改进DNAscope流程,加入Sentieon分型(Phasing)模块,高速准确分析PacBio HiFi数据进行SNP/Indel检测。
DNAscope LongReads运算效率高,速度相比开源软件有很大的提升。其中比对模块Sentieon Minimap2与原版相比提速2倍,而变异检测模块与DeepVariant相比提速6倍,有助于用户提升交付速度,降低计算成本。
准确度方面,DNAscope LongReads流程获得了FDA挑战赛PacBio数据的两个分项冠军,SNP的F1 score达到了0.9993,Indel为0.9943。在低深度下对比10x PB HiFi,16x PB HiFi,30x Illumina的全基因组测试结果,可以发现全基因组范围内16x的HiFi数据的准确率就已经超越了30x Illumina的数据,在低复杂度的基因组区域内即使10x的HiFi数据也可以超越Illumina的准确度。
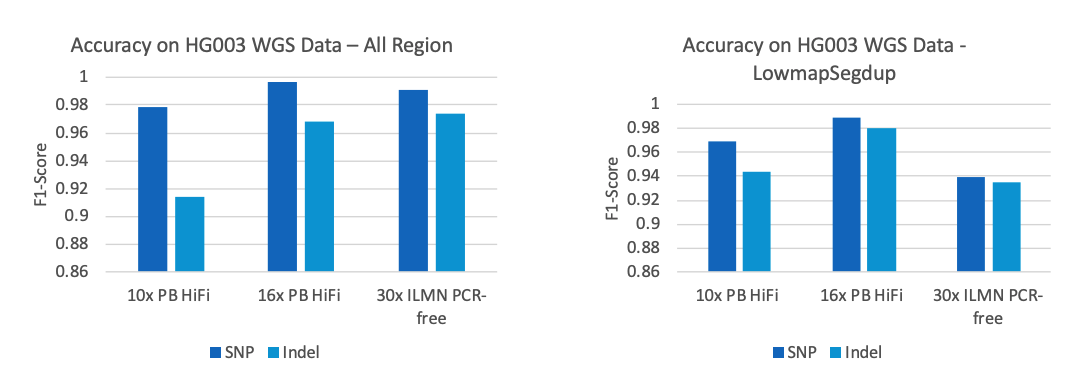
图5. SNP and Indel accuracy on HG003 WGS Data, PB HiFi vs ILMN
长读长数据可以覆盖Illumina序列所无法覆盖的区域,例如395个位于常染色体上复杂区域的临床相关基因(CMRG)。这些基因具有重要的临床价值,然而由于所处基因组区域较为特殊,短读长序列难以比对。以SMN1基因为例,该基因是脊髓性肌萎缩症的致病基因,最常见的突变是外显子7和8的缺失。从下图可以看出,只有PB HiFi数据可以覆盖相关区域,得出变异检测结果。
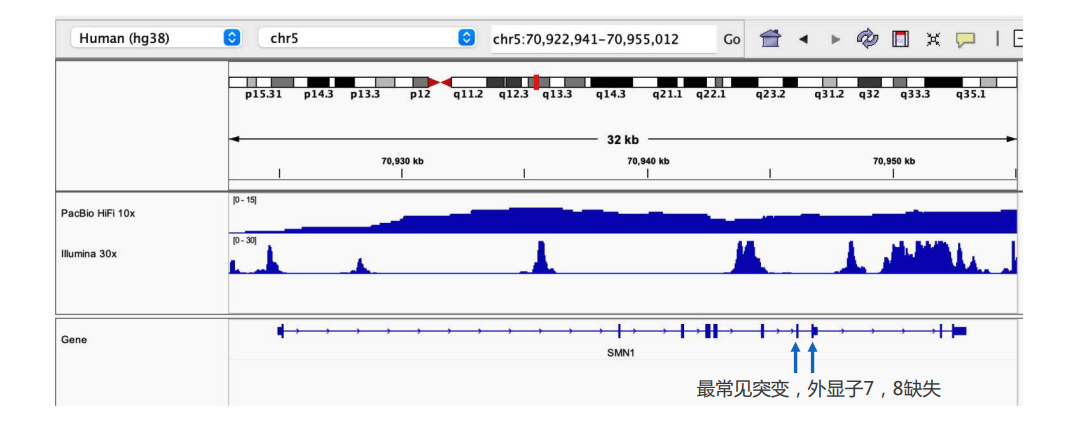
图6. PB HiFi covers SMN1 gene region when ILMN reads fail
通过结合双方在长读长变异检测流程中的特有优势,将点突变及大片段的结构变异整合分析,推出的完整HiFi全基因组重测序分析流程,将极大地加快PacBio HiFi测序的临床应用。基于本次成功经验,在未来,双方团队还会在Oxford Nanopore的数据处理上进行合作,为业界带来更多优质的Long Reads重测序的分析流程。同时双方也会持续在其他领域包括华大测序平台应用,以及“希望诊断计划”项目中保持合作,共同加速全基因组临床产品的市场教育和推广工作。
10月11日-13日,“天府健谈·CHS 2021第六届中国大健康产业升级峰会”召开,本次峰会由中国卫生信息与健康医疗大数据学会全科医学与健康管理工作委员会、亿欧EqualOcean主办,四川省卫生健康委、中国卫生信息与健康医疗大数据学会指导。
会上重磅发布了2021中国数字医疗企业百强榜系列榜单,经过历时数月的评选,拥有丰厚三代测序应用与发展的希望组凭借三代测序在临床医疗诊断产品上的不断创新,从众多企业中脱颖而出,成功入选“2021中国医疗创新力企业TOP10”。



![]()
希望组专注于在三代测序平台上进行技术开发与应用拓展,目前,已经形成了“科技服务”、“医学诊断”、“生物信息”、“先进制造”四大业务模块;在北京、武汉建设了合计超过5,000平米的科研服务平台、医学检验实验室和大数据分析数据中心,引进了国际先进的ONT PromethION 48、PacBio Sequel II、MGISEQ2000/T7、Bionano Saphyr光学图谱等技术平台。同时,希望组技术中心建立了完善的管理规章制度,为强化研发管理、激励技术创新,充分发挥公司技术中心在科技成果研发与转化方面作用提供机制和制度保障。



此次希望组被认定为“湖北省企业技术中心”,体现了政府主管部门对公司技术实力的高度认可,彰显了公司在技术研发和创新方面的实力。省级企业技术中心的建立是公司技术创新的又一里程碑,将成为公司今后高质量发展的助推器,推动公司技术研发创新水平的持续进步。
2021年9月23日, “安永复旦最具潜力企业2021”评选结果于上海揭晓。经过专家团队专业、严格的评选,希望组凭借三代测序大数据的技术应用,从众多参选企业中脱颖而出,当选为本年安永复旦最具潜力企业之一(种子企业)。
 希望组CEO汪德鹏(左二)现场领奖
希望组CEO汪德鹏(左二)现场领奖
“安永复旦最具潜力企业“评选活动由安永联合复旦大学管理学院举办,旨在于在企业的发展初期发掘出能在未来成为国家支柱产业的明日之星。该评选活动历经十一载,凭借其公开而权威的评选准则,已成为社会影响广泛、经济意义深远的活动。本届评选旨在发掘逆流而上、颠覆改革传统技术领域与进一步发掘市场空间的未来行业领袖,同时也希望能借此项活动,让资本市场更进一步认识中国优秀的新兴企业,助力其实现高速成长。

颁奖典礼祝酒合影
本次获奖是对希望组在三代测序应用开发领域工作的高度肯定,同时也为公司未来发展注入了强心剂。希望组前瞻性地看到了三代测序的产业级机会,未来将继续以科技服务为切入点,以生物信息为特色,结合医学研究服务与医学诊断,同时布局先进医疗制造领域,将自身发展深入基因测序的全产业链,从而推动基因测序产业的持续发展。

近日,由南方医学网、基因谷、早筛网共同举办的“2021基因检测行业发展高峰论坛”在广州成功举办。希望组作为中国长读长测序技术应用的开拓者,从参评企业中脱颖而出,荣获“2021基因检测行业年度最具投资价值企业”奖项!
会上,希望组CEO汪德鹏发表了题为“‘长枪短炮’进攻遗传病难题”的主题演讲,介绍了希望组在三代测序遗传病诊断领域的成果;同时提出了结合三代测序(长枪)和二代测序(短炮)技术,实现遗传病精准检测的方案。希望组致力于三代测序技术的应用,在三代测序领域深耕多年,构建了ONT PromethION 48、PacBio Sequel II三代测序技术平台;基于三代测序,建立了人类基因组结构变异数据库(dbSV),开发了结构变异分析软件(GrandSV)。此次推出的“‘长枪短炮’方案,利用二代测序低成本、高通量、高准确度的特点,用于基因编码区小变异(SNP/Indel)的检测;结合三代测序长读长优势,通过长读长测序全基因组测序,利用希望组分析软件和数据库实现结构变异的精准检测,有效提升遗传病的检出率,可以发现新的疾病相关结构变异断点,为遗传病诊断、辅助生殖和基因治疗提供了新的思路。

希望组CEO汪德鹏发表主题演讲
在随后的圆桌分享环节中,就“全面建设健康中国与基因检测未来展望”的主题,公司CEO汪德鹏与专家及企业代表共同探讨中国基因检测行业的现状,未来发展,以及三代测序的应用方向和前景。

希望组CEO汪德鹏参与圆桌会议
希望组将继续践行“让生命充满希望”的使命,坚守“ 创新、创造价值,并创造利润”经营原则,不断探索长读长测序技术在科学研究及医学诊断领域的应用,推动新技术的临床转化和落地,为健康中国建设做出积极贡献,实现全球三代测序技术与应用领域领军企业的战略目标。
2021年9月6日,北京希望组生物科技有限公司(以下简称“希望组”)正式引进深圳华大智造科技股份有限公司(以下简称“华大智造”)旗下超高通量测序仪DNBSEQ-T7,共同推进遗传病精准诊断技术的创新与市场化落地。

“长枪短炮”模式创新
瞄准遗传病精准诊断
高通量测序技术近年来在遗传病检测领域得到了广泛应用,以靶向测序及全外显子组测序为代表的技术已成为遗传病诊断的重要工具。全基因组测序(WGS)理论上可以同时检测单核苷酸变异、结构变异(含拷贝数变异)及线粒体变异等,有望进一步提升临床检测的效能。
作为国内长读长、高通量测序服务的探路者,希望组在遗传病检测领域积累了丰富的项目经验。基于长读长测序技术(Long Reads Sequencing, LRS),希望组解决了大片段结构变异检测难题,让面肩肱型肌营养不良症、杜氏肌营养不良症等疑难基因组病的精准诊断不再遥不可及。值得关注的是,希望组在业界首开先河,基于现有方案结合DNBSEQ-T7超高通量、高准确性的技术优势,提出了“短读长+长读长”遗传病精准诊断新模式,进一步提高致病变异的检出率,让更多的遗传病患者得到确诊。随着DNBSEQ-T7等强势机型的陆续入驻,希望组将在遗传病精准诊断领域开创更多可能性。
提高检测通量、降低检测成本
推进遗传病精准诊断市场化落地
“DNBSEQ-T7的顺利入驻是继2019年希望组引进MGISEQ-2000之后对高通量测序服务能力的又一次重大升级和重要战略部署,”希望组CEO汪德鹏表示,“希望组始终致力于将基因测序技术创新性应用于科技服务、精准医学领域。随着DNBSEQ-T7超高通量测序平台的启用,希望组将成为同时拥有长、短读长超高通量测序平台的服务商,为用户提供更快、更丰富、更优质的服务!”

华大智造首席运营官蒋慧表示,“高质量测序的核心工具平台是生命科学研究、精准医疗以及相关产业创新发展的基础。我们希望通过DNBSEQ-T7测序平台的技术优势,助力希望组的核心业务实现突破性跃迁,从而共同推动生命科学及精准医学领域的创新发展,赋能更多下游用户。”

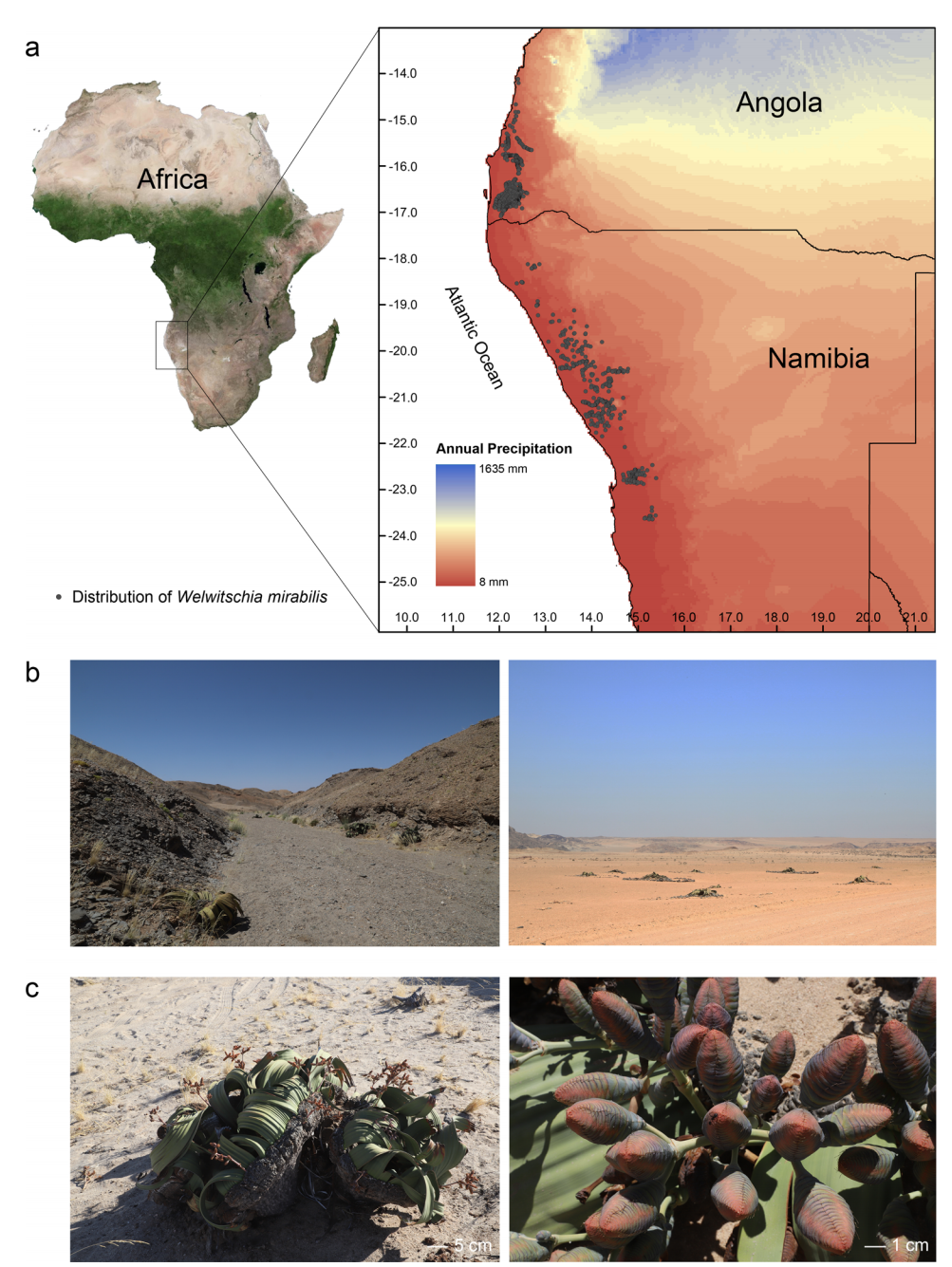
图1,百岁兰形态、生境及其分布
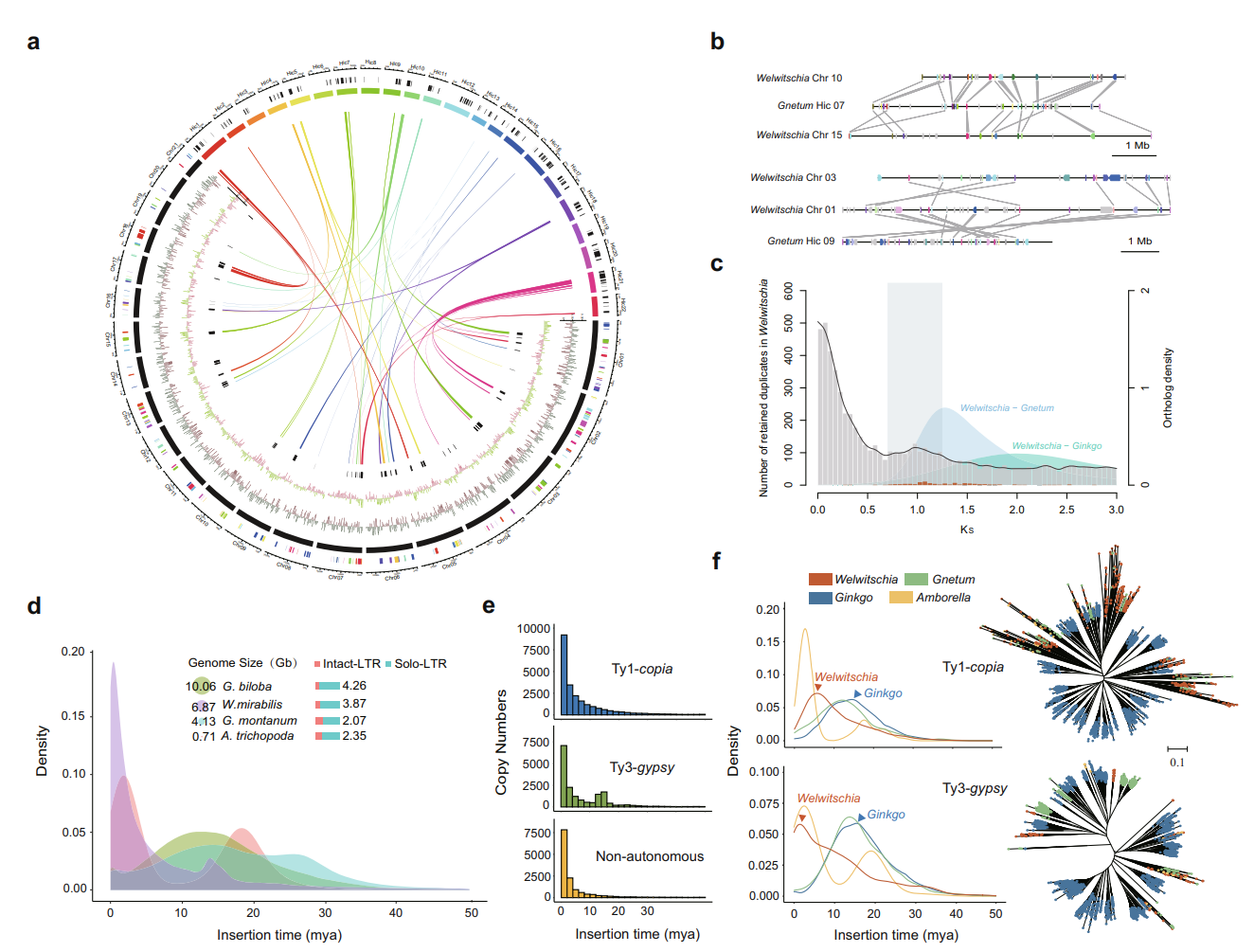
图2. 百岁兰基因组演化动态和历史
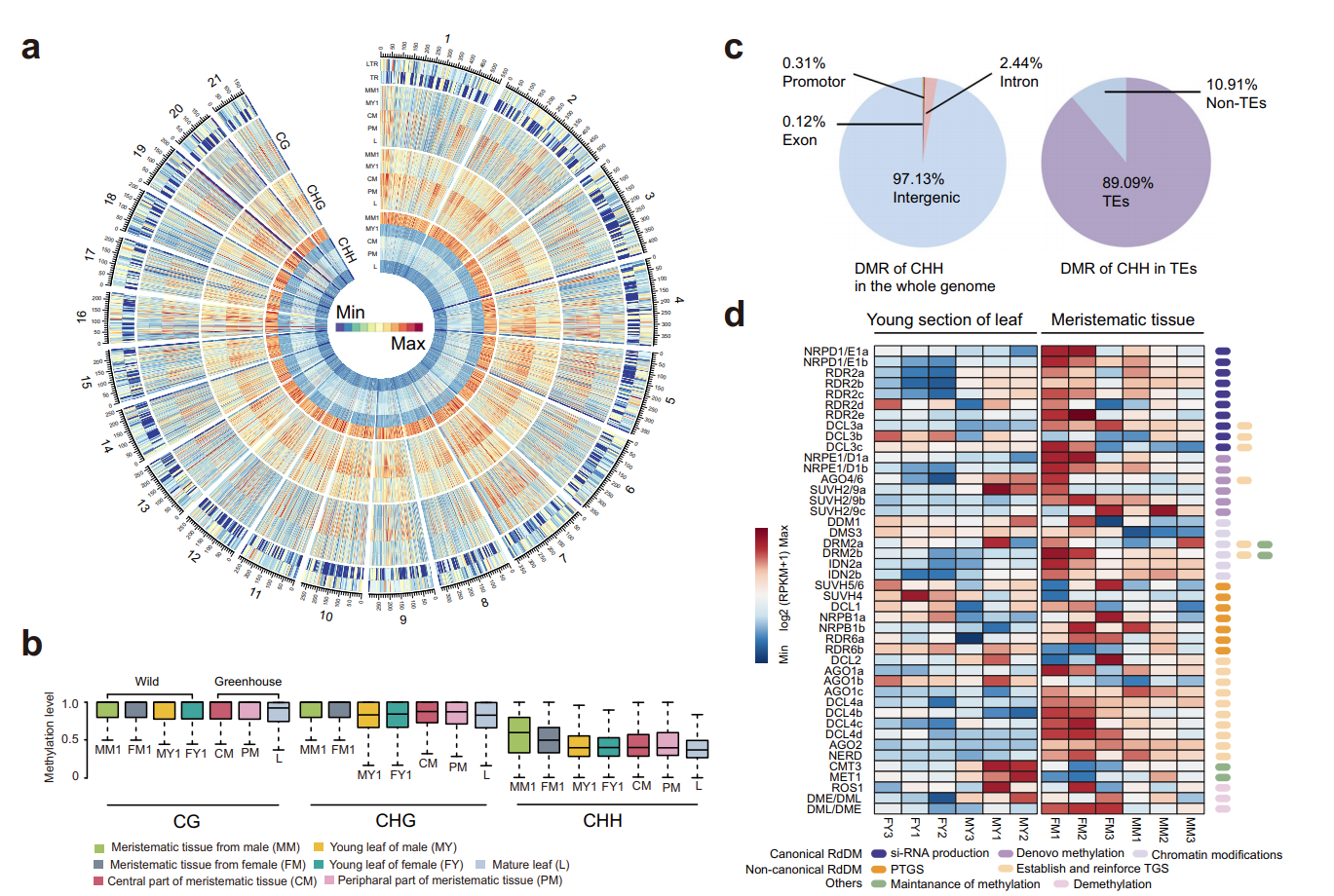

图3\4. 百岁兰甲基化图谱,重度甲基化百岁兰基因组脱氨基导致的低GC
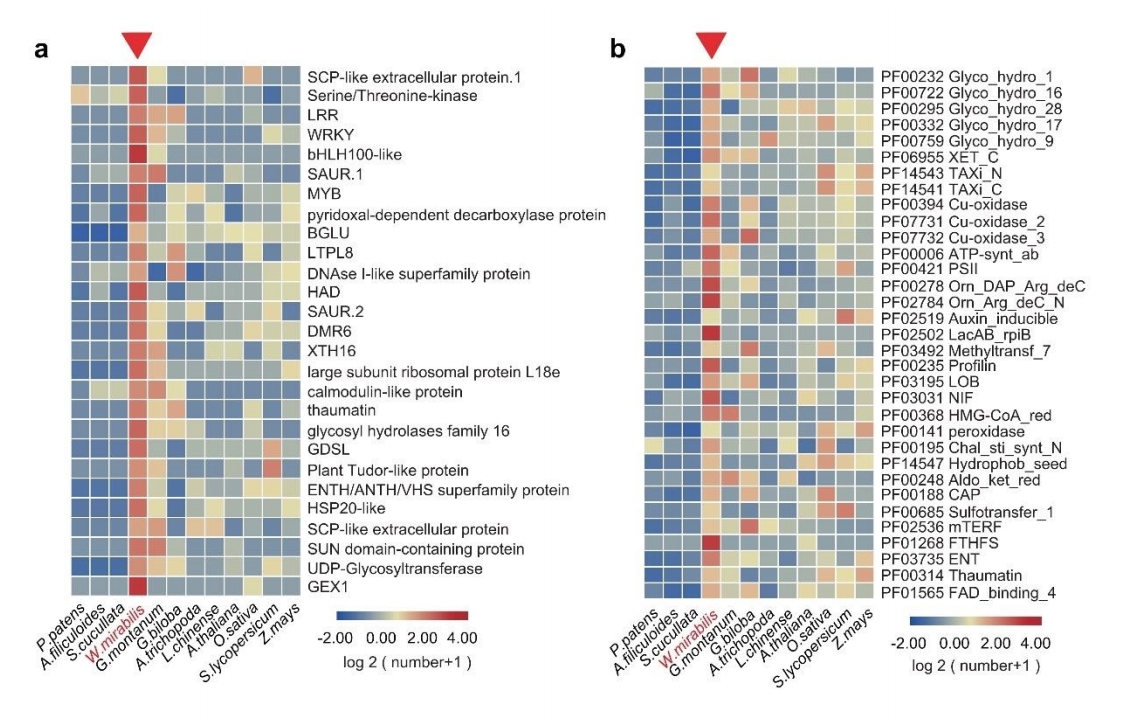

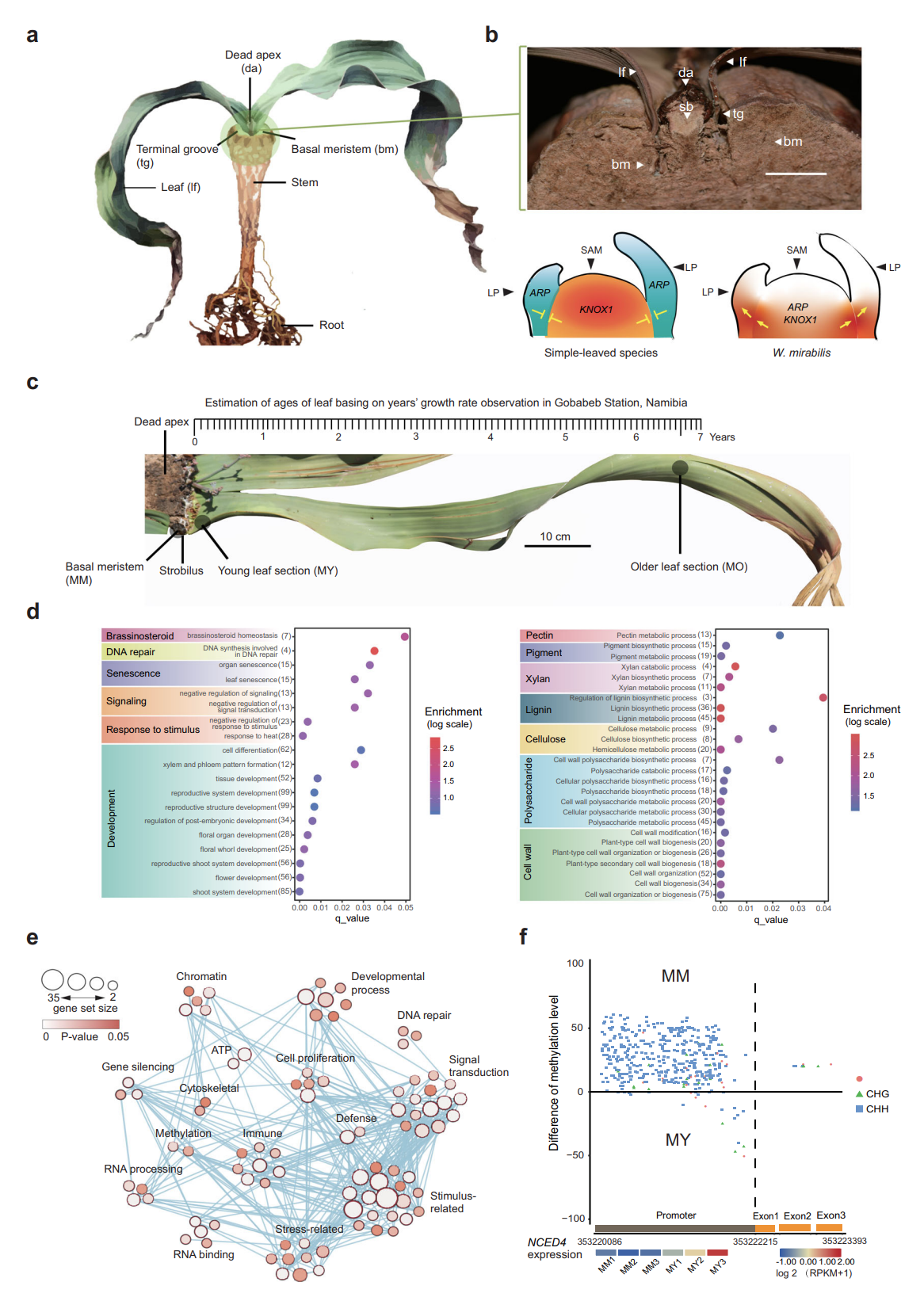
图5/6/7. 百岁兰特性扩张基因家族及新老叶段基因家族的差异表达

| 武汉希望组医学检验实验室有限公司 危险废弃物产出明细公示表(2020年度) | |
| 组织机构代码 | 91420100MA4KXQ2J1P |
| 法定代表人 | 汪德鹏 |
| 生产地址 | 武汉市东湖新技术开发区花城大道8号武汉软件新城C11栋17楼 |
| 生产经营内容 | (共1个一级诊疗科目)医学检验科(临床免疫、血清学专业,临床细胞分子遗传学专业) |
| 危险废弃物类别 | HW01医疗废物 |
| 危废名称 | 感染性医疗废物 |
| 危废代码 | 841-001-01 |
| 处置方式 | Y10医疗废物焚烧 |
| 主要成分 | 废弃离心管、废吸头、废试剂瓶和废采血管,一次性医用口罩、手套、帽子等实验用品 |
| 产废工序 | 一次性医用口罩、手套、帽子等实验用品为检验人员使用后医疗废物,废弃离心管、废吸头、废试剂瓶和废采血管为接触过样本或检验试剂的废弃医疗废物。 |
| 危险特性 | 潜在感染性 |
| 安全措施 | 我机构产生的医疗废物统一使用专用医疗垃圾袋密封,经次氯酸钠喷洒、高温高压等消毒措施处理后集中存放于医疗废物暂存间医疗垃圾桶。医废暂存间由专人进行管理,暂存环境使用次氯酸钠和紫外灯照射消毒。我机构医疗废物委托给武汉汉氏环保工程有限公司处置。 |
| 贮存区域 | 医疗废物暂存间 |
| 处置去向 | 武汉汉氏环保工程有限公司 |
| 医废暂存间管理员 | 杨婷婷15012731512 |
| 紧急联系人 | 全伟鹏15927151043 |
| 相关联系人 | 刘兰兰18071062020 |
| 产生量(2020年)(单位:吨) | 6.2 |
| 转运量(2020年)(单位:吨) | 6.2 |
| 库存量(2020年)(单位:吨) | 0 |

希望组

希望组科技服务

希望组诊断服务