项目文章丨兰州大学从全基因组水平揭示象草花青素积累和快速生长分子机制
近日,兰州大学草地农业科技学院联合广西畜牧研究所及国际家畜研究所共同合作的象草基因组研究成果以“The elephant grass (Cenchrus purpureus) genome provides insights into anthocyanidin accumulation and fast growth”为题在国际知名期刊《Molecular Ecology Resources》(3年IF=7.15)在线发表。希望组科技服务为本研究提供了Illumina、Nanopore和Hi-C测序服务,承担了基因组的组装及注释任务。该研究首次报道了象草的高质量染色体级别基因组,明确了象草的进化地位,在基因水平解析了紫色品种象草 “紫色”花青素积累的机制,并提出C4光合作用和激素信号转导通路的扩张可能有助于象草快速生长的新见解[1]。

象草(Cenchrus purpureus Schumach)因大象爱采食而得名,是禾本科、黍族多年生大型草本植物,原产于亚洲。象草因其具有生物量大、生长快速、适应性强等特点,被用作重要的饲草作物在全世界热带及亚热带被广泛种植。此外,由于象草在生物能方面的优势也使其潜在的能源草。该研究是对象草研究的重大突破,为象草进化、性状改良和功能基因研究提供了理论基础。

图1 紫色象草
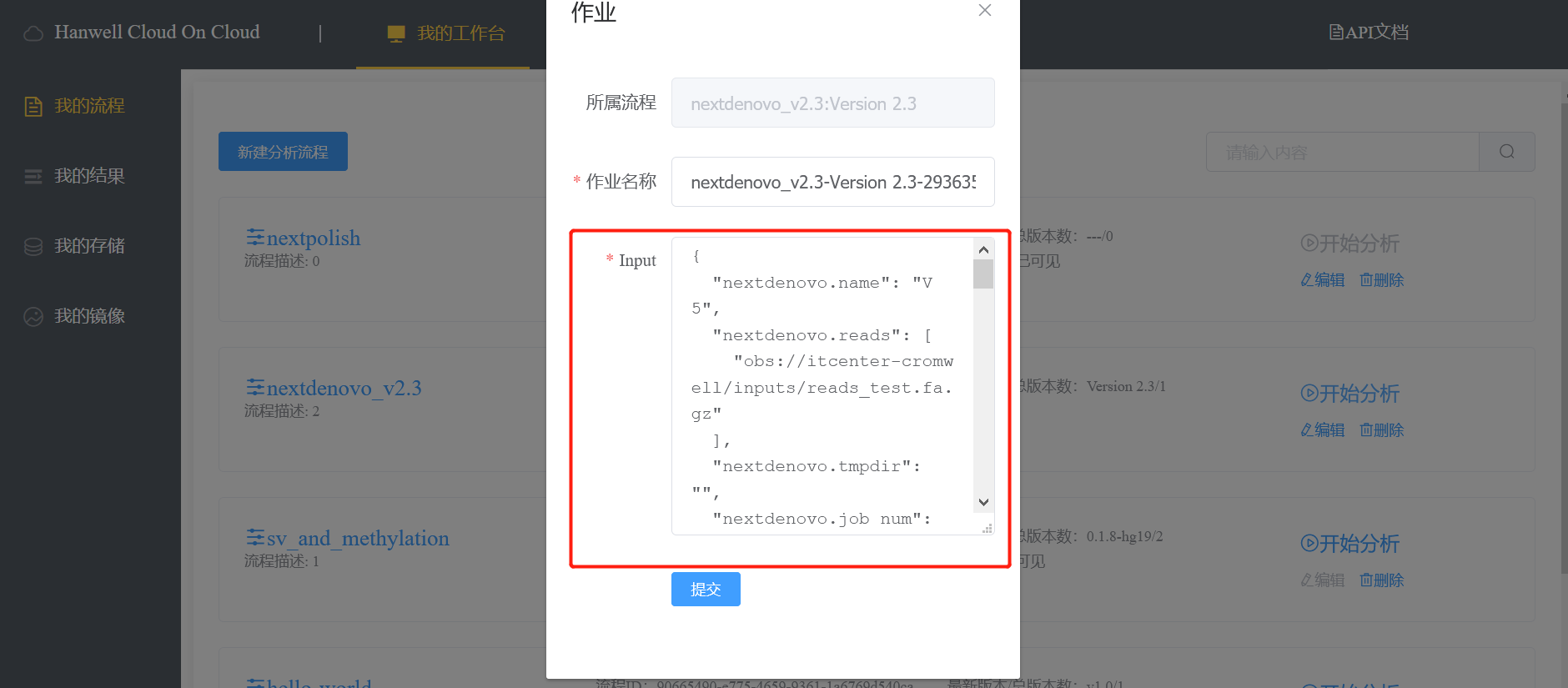
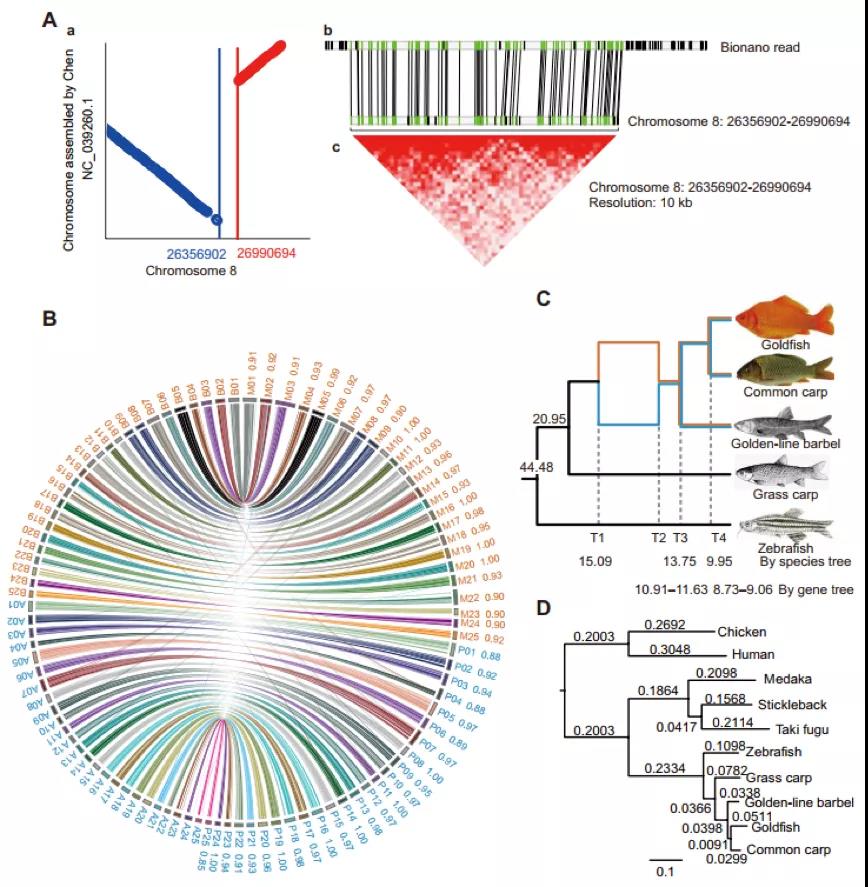
研究团队以紫色象草(Cenchrus purpureus cv. Purple)为材料,K-mer评估显示象草具有较高杂合(1.5%)。利用Illumina、Nanopore、Hi-C测序。采用NextDenovo + SMARTdenovo策略组装获得1.97Gb的基因组, Contig N50 为1.83Mb,最长Contig达到15.1Mb。结合Hi-C数据对基因组辅助染色体挂载及遗传连锁图谱,得到14条染色体,挂在率为96.65%。BUSCO评估结果达 97.8%,预测注释基因65,927个。
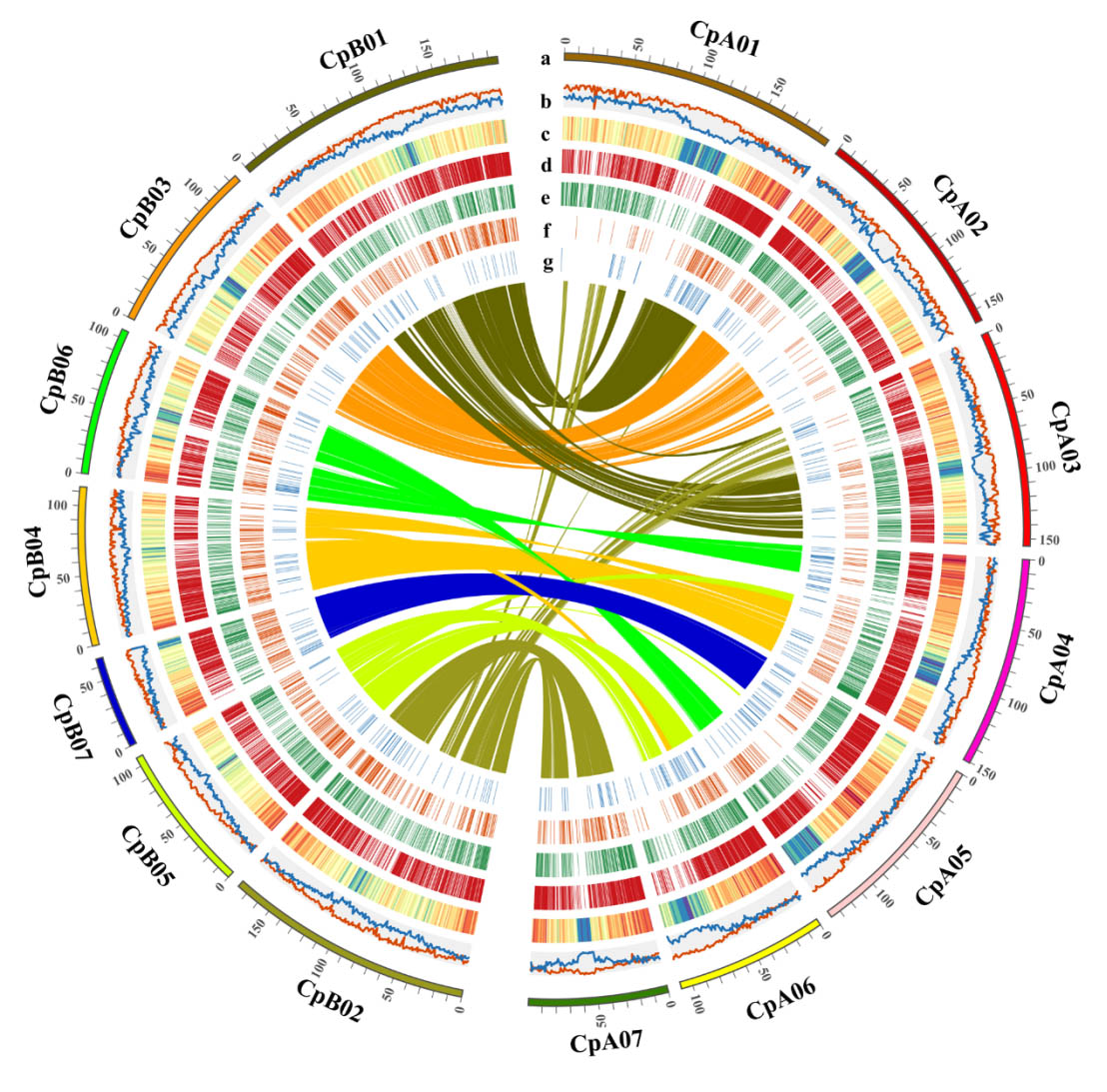
图2 象草亚基因组特征
象草为异源四倍体(2n=4x=28),包含A’和B两个亚基因组。研究表明同属二倍体植物珍珠粟(Cenchrus americanus,2n=2x=14)的A基因组与象草A’基因组具有更高同源性。通过共线性分析研究者成功将象草的A’和B两个亚组区分开来,并利用单拷贝基因分析证明象草A’亚基因组和珍珠粟A基因组具有较近的同源性。象草A’A’BB的异源四倍体基因组大约起源于6.61 (4.11-10.92)MYA,并发生了较大的染色体重组。此外,研究者还利用转录组分析了象草亚基因组显性表达,结果表明其可能行使不同的功能。

图3 紫色象草花青素积累机制
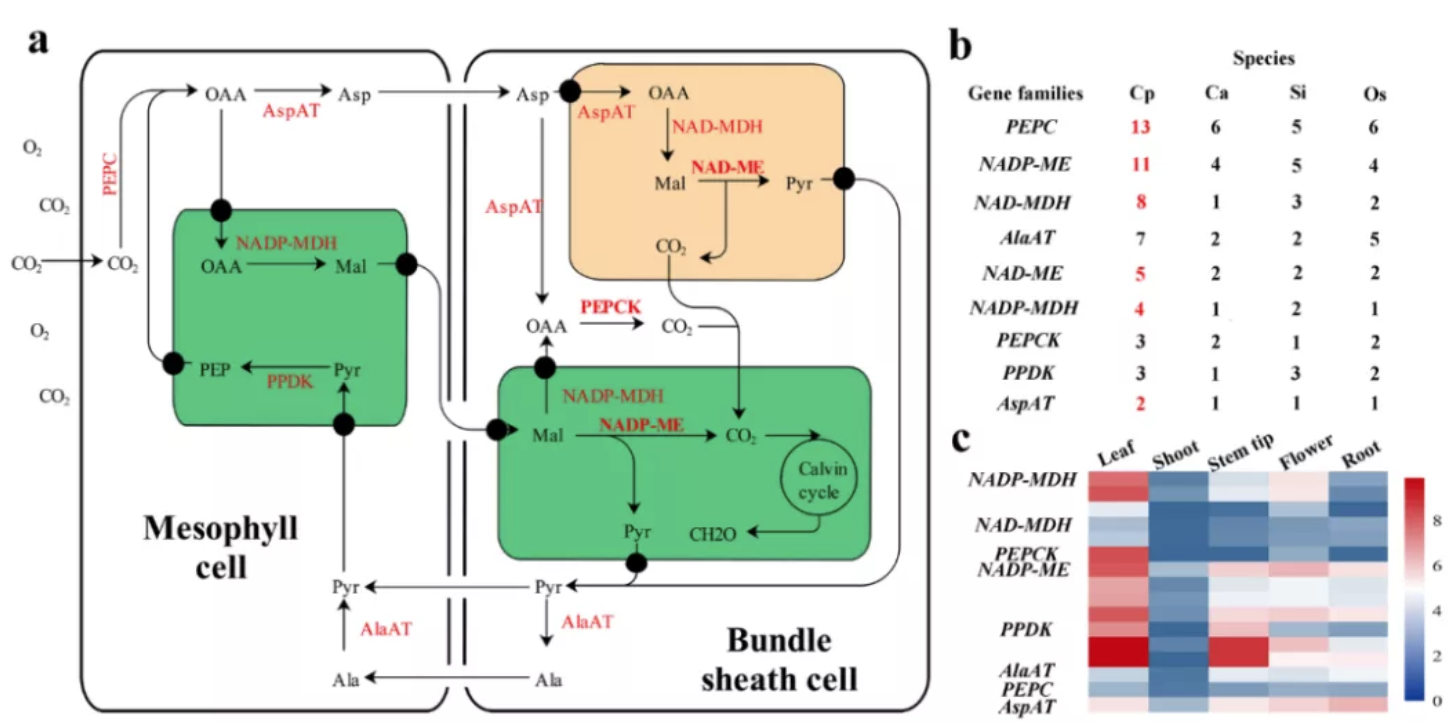
图4 象草C4光合途径
该研究利用报道的高质量的象草基因组、解析了花青素合成及快速生长机制,为象草作为优良饲草和潜在能源草的分子改良育种具有重要意义。此外,对于该属的进化以及其它物种的开发利用提供了重要资源。兰州大学草地农业科技学院张吉宇教授为通讯作者、广西畜牧研究所易显凤研究员、国际家畜研究所Jones Chris博士为共同通讯作者。兰州大学草地农业科技学院博士生闫启为第一作者、团队博士生吴凡、许攀和希望组孙宗毅为共同第一作者。
1. Yan Q, Wu F, Xu P, Sun ZY, Li J, Gao LJ, Lu LY, Chen DD, Muktar M, Jones C, Yi XF, Zhang JY. The elephant grass (Cenchrus purpureus) genome provides insights into anthocyanidin accumulation and fast growth. Mol Ecol Resour 2020, doi:10.1111/1755-0998.13271