
硬核爆发-希望组2020三代基因组文章集锦-植物篇
高质量基因组揭示棉花A亚基因组起源[1]
影响因子:25.455
发表日期:2020.04.13
三代测序平台:PacBio RSII&Sequel
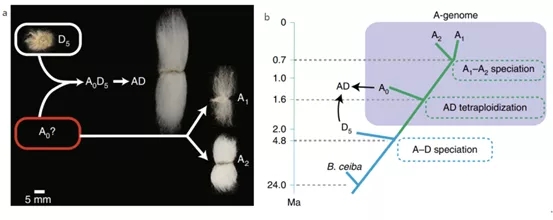
图1 棉花A基因组起源与进化模型(a)和重要进化事件(b)
ONT测序助力攻克首个高质量角苔参考基因组[2]
影响因子:13.297
发表日期:2020.02.10
三代测序平台:Nanopore PromethION
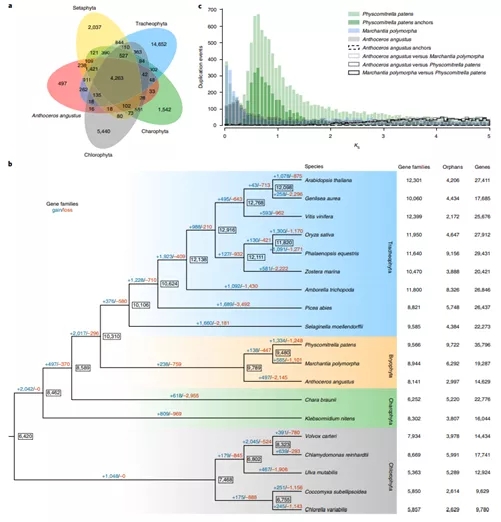
图2 芽胞角苔与18个绿色植物的比较基因组分析。a, 基于OrthoMCL的基因家族聚类比较。b, 19个绿色植物的基因家族获得(+)/丢失(-)情况比较,红框标注苔藓类群分支。c, 芽胞角苔、小立碗藓和地钱的全基因组加倍事件分析。
高质量油桐基因组,荣登GPB期刊“封面故事”[3]
影响因子:6.597
发表日期:2020.04.07
三代测序平台:PacBio RSII
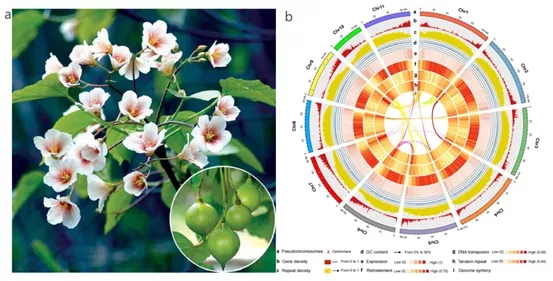
图3 油桐(a)及其基因组景观(b)
白木香—瑞香科第一个染色体水平基因组[4]
影响因子:4.688
发表日期:2020.03.02
三代测序平台:Nanopore GridION
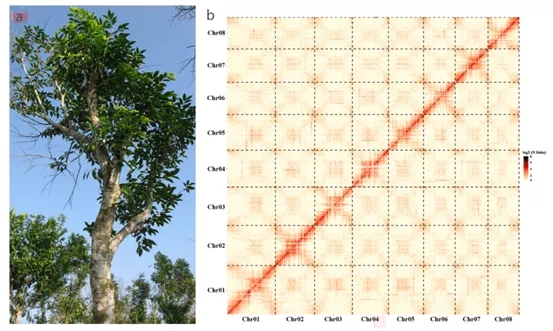
图4 白木香(a)及其基因组Hi-C热图(b)
园艺观赏植物文竹染色体水平基因组[5]
影响因子:3.368
发表日期:2020.04.01
三代测序平台:Nanopore GridION
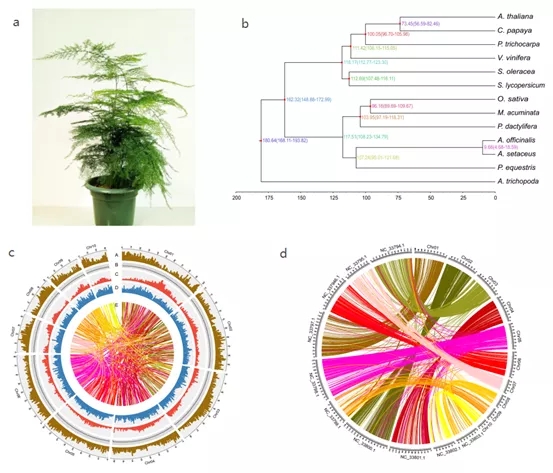
图5 文竹高质量基因组。(a)文竹。(b)基于1002个单拷贝直系同源基因的系统发生树。(c)文竹基因组景观。(d)文竹与芦笋基因组线性比较
影响因子:4.688
发表日期:2020.02.26
三代测序平台:Nanopore GridION
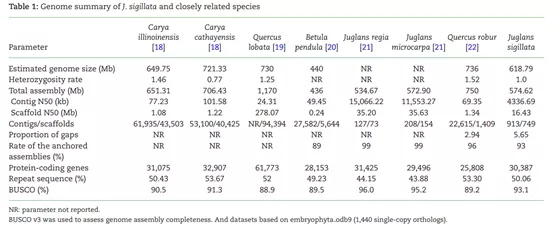
图6 铁核桃与其近源种基因组比较
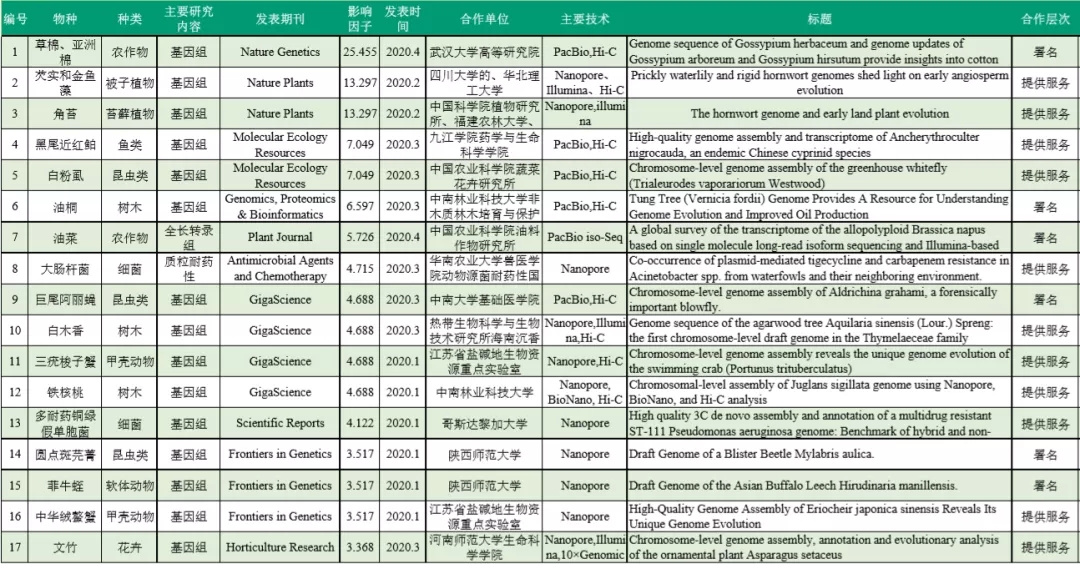 ▼
▼参考文献:
1. Huang, G. et al. Genome sequence of Gossypium herbaceum andgenome updates of Gossypium arboreum and Gossypium hirsutumprovide insights into cotton A-genome evolution. Nat Genet (2020)doi:10.1038/s41588-020-0607-4.
2. Zhang, J., Fu, X., Li, R. et al. The hornwort genome and early landplant evolution. Nat. Plants 6, 107–118 (2020).https://xs.scihub.ltd/https://doi.org/10.1038/s41477-019-0588-4
3. Zhang, L. et al. Tung Tree (Vernicia fordii) Genome ProvidesA Resource for Understanding Genome Evolution and Improved Oil Production.Genomics, Proteomics & Bioinformatics S167202291830216X (2020)doi:10.1016/j.gpb.2019.03.006.
4. Ding, X. et al. Genome sequence of the agarwood tree Aquilariasinensis (Lour.) Spreng: the first chromosome-level draft genome in theThymelaeceae family. GigaScience 9, giaa013 (2020).
5. Li, S.-F. et al. Chromosome-level genome assembly, annotation andevolutionary analysis of the ornamental plant Asparagus setaceus. HorticRes 7, 48 (2020).
6. Ning, D.-L. et al. Chromosomal-level assembly of Juglanssigillata genome using Nanopore, BioNano, and Hi-C analysis. GigaScience 9,giaa006 (2020).
让生命充满希望!——“希望组”“未来组”品牌整合
疫情之后,一切都改变了。
也许这是一个要改变人类生活方式,生产方式,合作方式的“大历史”事件。

2020年4月13日于武汉
武汉希望组医学检验实验室满分通过全国新型冠状病毒核酸检测室间质量评价
近日,国家卫生健康委临床检验中心公布全国新型冠状病毒核酸检测室间质量评价结果,武汉希望组医学检验实验室满分通过,并获得室间质评合格证书。

关于全国新型冠状病毒核酸检测室间质量评价
核酸检测是新型冠状病毒肺炎(Corona Virus Disease 2019,COVID-19)确诊的重要手段,为了解我国新型冠状病毒(2019 novel coronavirus, 2019-nCoV;国际病毒分类委员会将2019-nCoV命名为SARS-CoV-2)核酸检测的开展现状及质量状况,帮助临床实验室发现检测中存在的问题并进行改进,使得核酸检测在疾病防控工作中更好地得到应用,国家卫生健康委临床检验中心于2020年3月开展了新型冠状病毒核酸检测室间质量评价。本次“全国新型冠状病毒核酸检测室间质量评价”主要评价实验室对新型冠状病毒核酸检测的能力,重点考察实验室检测的分析性能,包括分析敏感性和分析特异性。
室间质量评价(EQA,external quality assessment),是多家实验室分析同一标本、并由外部独立机构收集和反馈实验室上报的结果、以此评价实验室操作的过程。通过实验室间的比对判定实验室的校准、检测能力以及监控其持续能力。室间质量评价是国际公认的临床实验室全面质量管理的重要组成部分,也是世界上多数国家临床实验室行政管理和实验室认可的基本要求。
武汉希望组医学检验实验室满分通过全国新型冠状病毒核酸检测室间质量评价,证明了武汉希望组新冠病毒核酸检测结果的可靠性、稳定性,有能力为一线抗疫提供高效精准的核酸检测筛查。同时,武汉希望组正在开发基于纳米孔测序技术的冠状病毒全基因组检测技术,与基于qPCR的技术形成有力互补,为疫情防控提供关键技术支持!
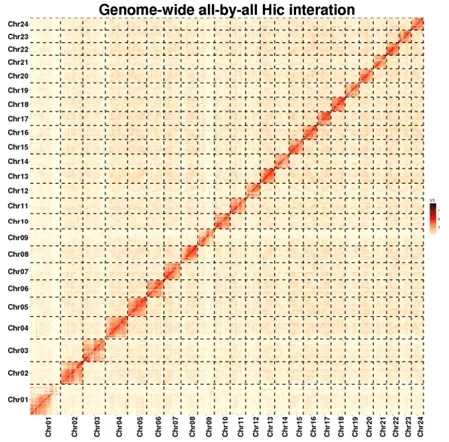
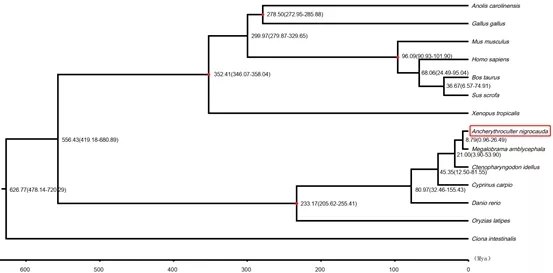
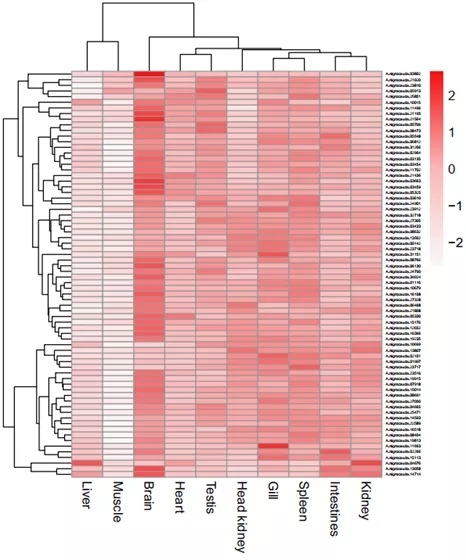
项目文章||九江学院张化浩博士研究团队发表中国特有物种黑尾近红鲌高质量基因组


希望组与Bionano合作开展COVID-19感染患者基因组变异研究
Bionano 将用于鉴定导致冠状病毒疾病易感性的基因变异的研究
第一项研究将在中国武汉进行;欧洲和北美的研究有望紧随其后

*本文基于Bionano Genomics公司官方网站新闻”Bionano Genomics Will Be Used in Research Identifying Gene Variantsthat Contribute to Coronavirus Disease Susceptibility”翻译。
项目文章两连发||三代测序助力药用动物圆点斑芫菁、菲牛蛭基因组草图组装
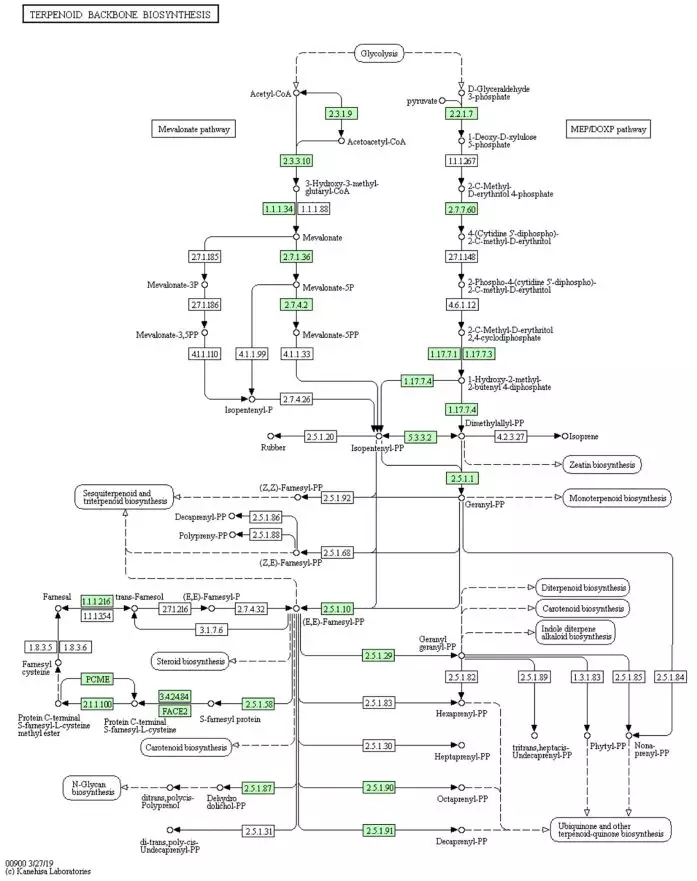
图1 萜类生物合成“KEGG通路图”,绿色方框基因在圆点斑芫菁基因组中发现。
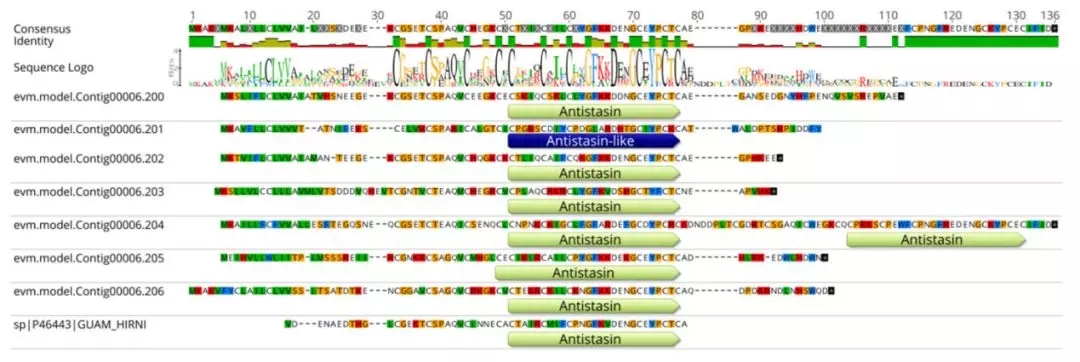
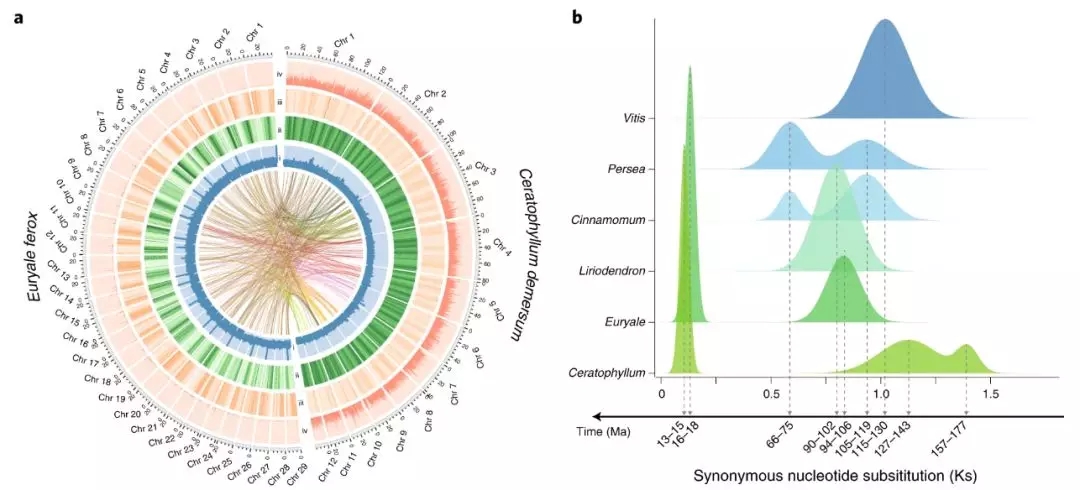
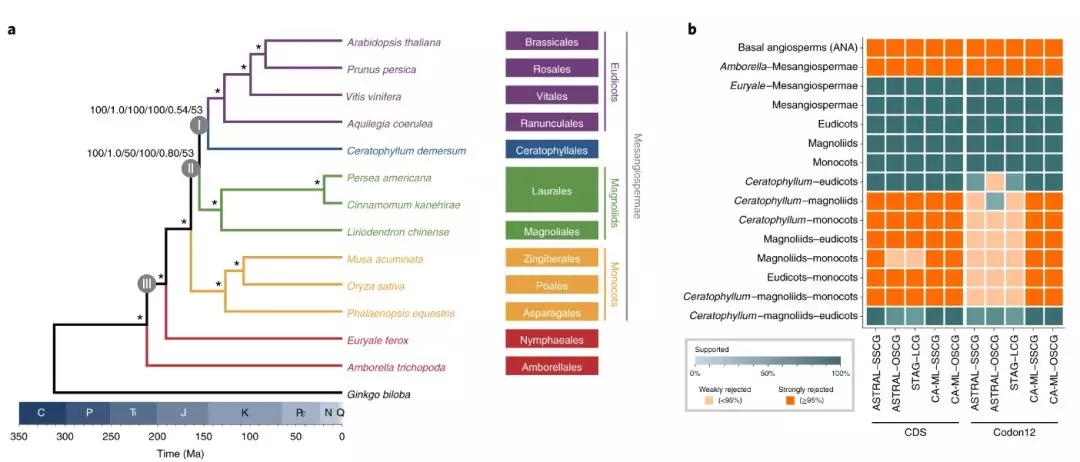
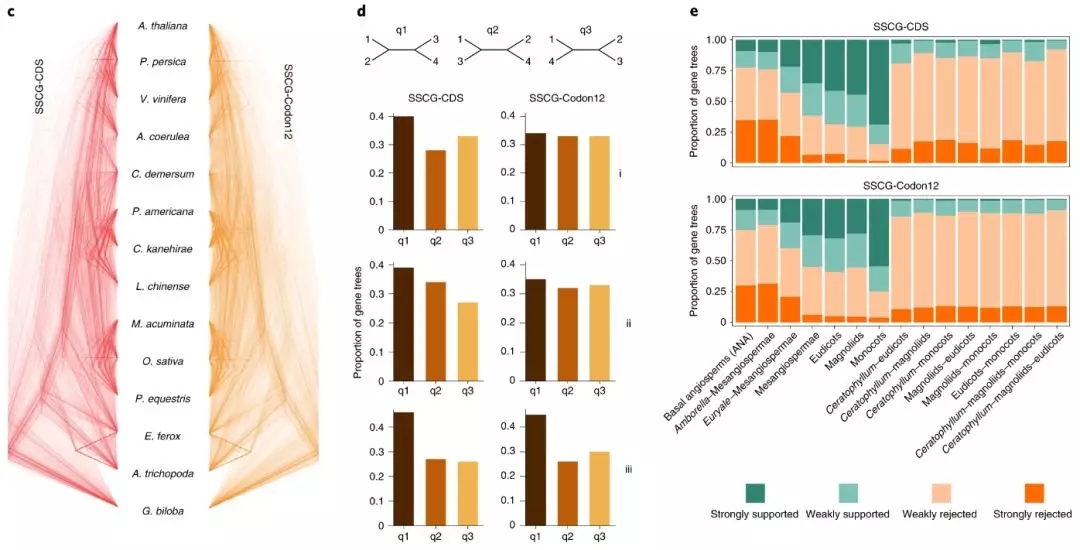
项目文章||芡实与金鱼藻三代基因组揭示早期被子植物演化
官方微信公众号

希望组

希望组科技服务

希望组诊断服务