项目文章|Nanopore测序破译栽培桑树基因组,解决桑树物种分类、染色体组倍性争议,揭示湖桑起源之谜
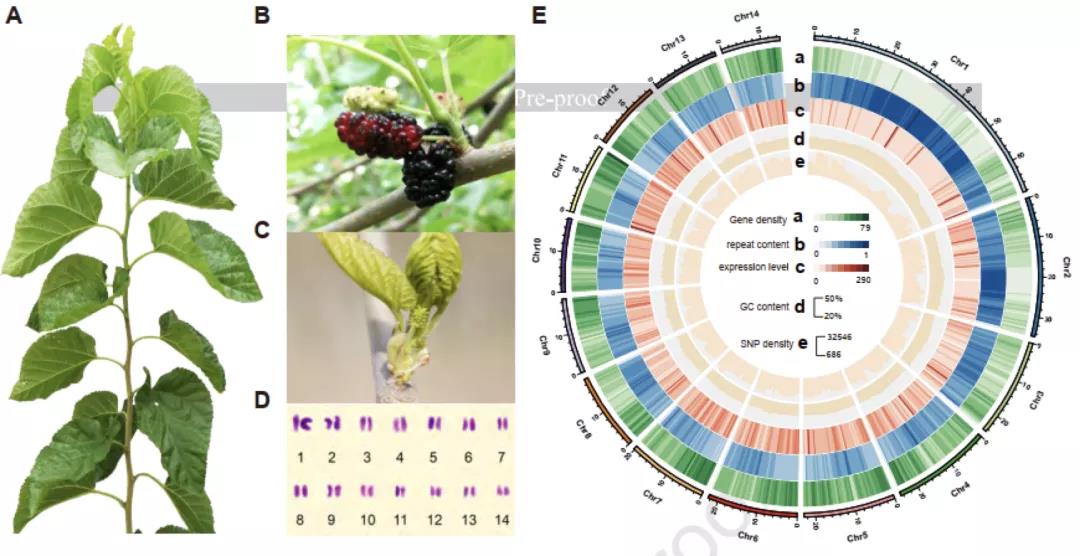
图1 “荷叶白”的植物形态、核型分析和基因组组装结果。
研究者选择栽培桑树“荷叶白”(又名湖桑32号)为研究对象(图1ABC)。核型分析表明,栽培桑树“荷叶白”的体细胞有丝分裂过程和花粉母细胞减数分裂过程中,28条染色体形成规则的14对二价体(图1D)。利用Nanopore+短读长+Hi-C策略进行基因组测序和组装,最终获得了基因组大小为346.39 Mb,scaffold N50为22.87 Mb的栽培桑树基因组(图1E)。利用该高质量基因组进行系统发育树构建,发现野生川桑和栽培桑树分化时间已有10.1个百万年(图2A)。与葡萄和桃树基因组共线性分析发现,栽培桑树基因组除了具有双子叶植物共有的γ古六倍化事件之外,没有新的全基因组加倍(WGD)事件发生。因此,栽培桑树基因组为二倍体,并非来源于野生川桑基因组的同源或异源加倍。
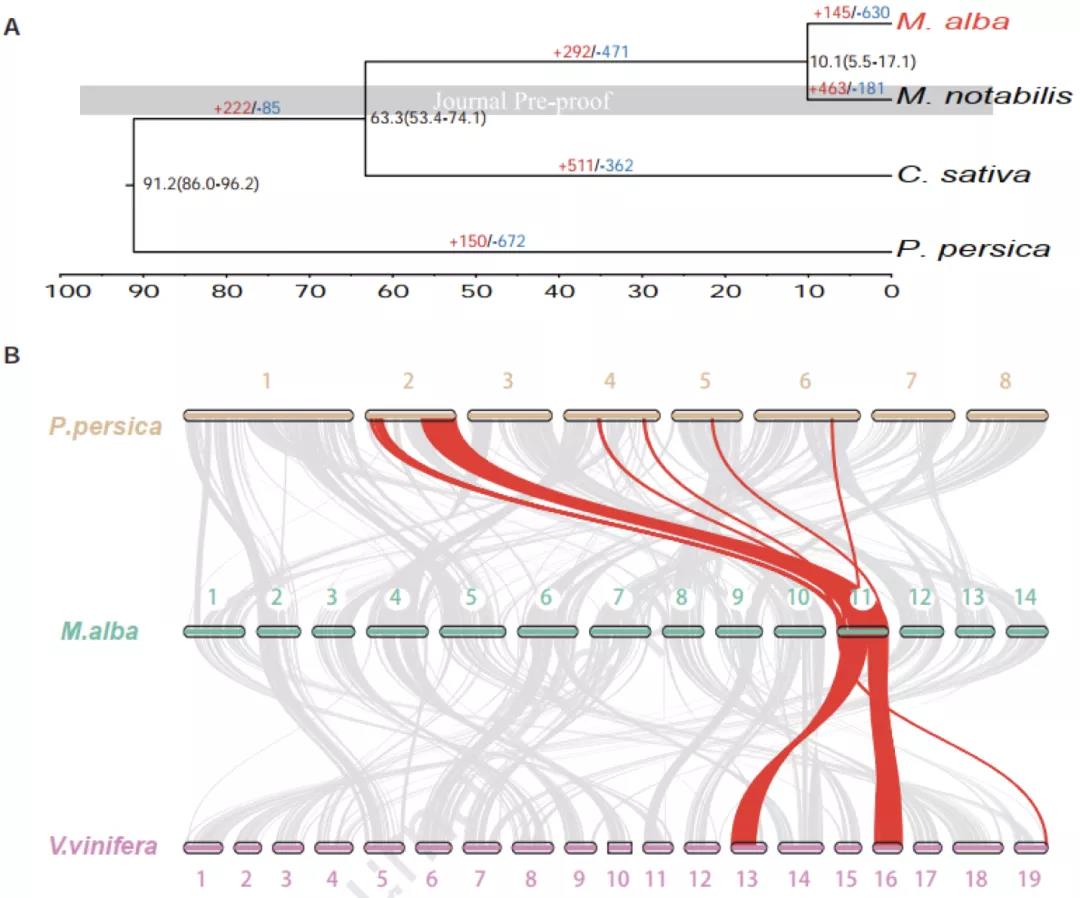
图2 (A)白桑(Morus alba)与川桑(M. notabilis)的分化距离在~10.1个百万年左右,(B)白桑与葡萄(Vitis. vinifera)和桃(Prunus persica)基因组共线性分析。
现有栽培桑树按照形态学特征分为白桑、鲁桑、山桑、广东桑和瑞穗桑五个种,并不能真实反映桑树品种之间的系统发育关系。本研究收集了132分栽培桑树种质(除广东桑外)进行重测序,获得了14.27Mb的单核苷酸多态性(SNP)数据,利用该数据构建系统发育树,没有得到与形态分类相似的聚类结果,在分子水平将白桑、鲁桑、山桑和瑞穗桑这4种栽培桑树种鉴定为同一物种,即白桑(Morus alba L)。
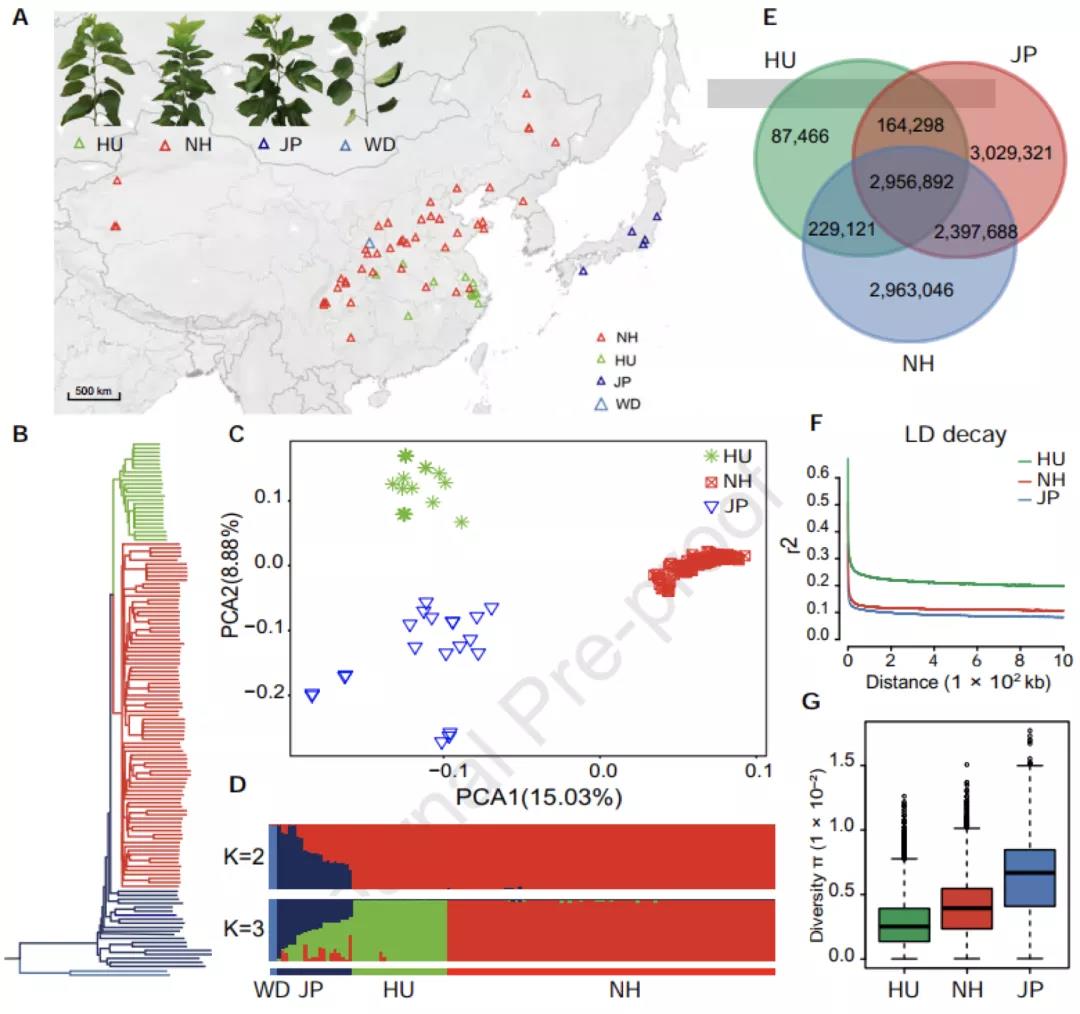
图3 134份桑树种质资源的群体结构、核酸多样性分析
群体结构分析将134份栽培桑树种质划分为三个大群:中国湖桑群体,中国北方和西南群体,日本群体(图3A)。系统发育和主成分分析均表明中国桑树群体与日本桑树群体遗传距离较远,湖桑与来自于北方和西南地区的桑树具有明显的分化距离(图3BCD)。遗传多样性分析显示,湖桑的遗传多样性只有其他群体的一半,有强烈的人工选择痕迹。因此,太湖流域的湖桑与其他桑树群体在更早时期就已分开,成为一个独特的品种支系。同时自唐代以来,我国桑蚕业核心区域南移,湖桑作为独立种质资源受到了江南人民持续有目的的选育。这与崧泽遗址的孢粉学研究和吴兴钱山漾考古学证据可以相互印证。