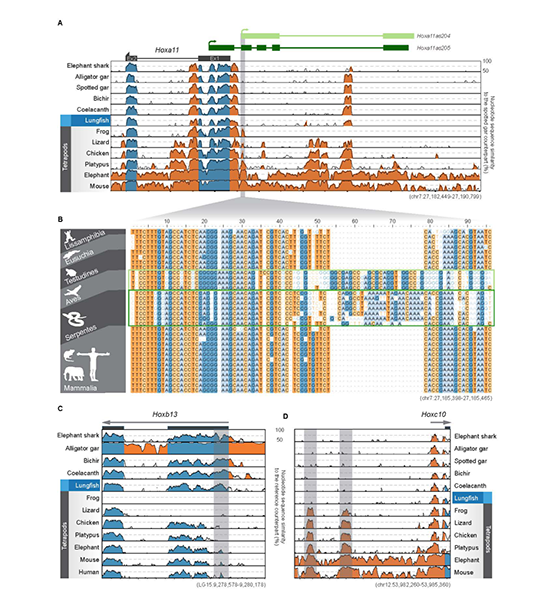
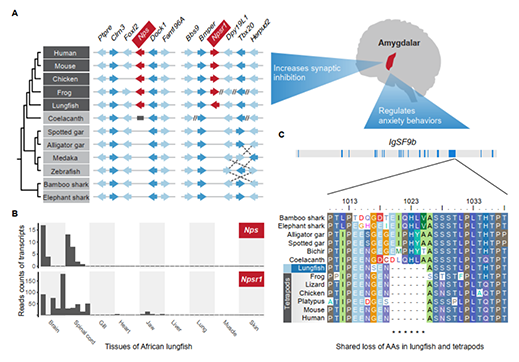
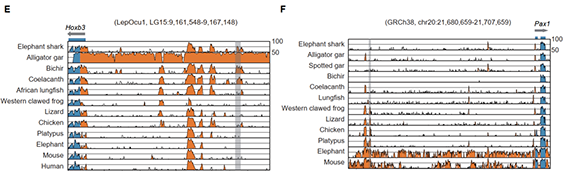
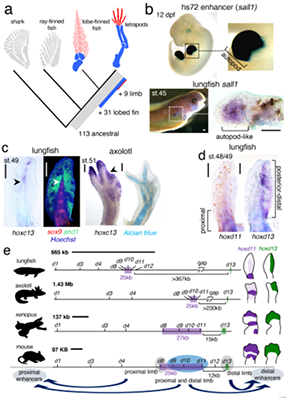
2009年首个单细胞转录组测序技术问世,开启了单细胞组学时代(scRNA-seq)(Tang et al., 2009)。过去十余年间单细胞测序技术的不断发展极大地加速了生物医学领域的相关研究,帮助科研人员克服了稀有生物样本以及生物样本内生异质性等重大挑战,一系列模式生物及人类自身的单细胞转录组图谱也由此诞生。然而目前的单细胞测序技术几乎全都是基于二代测序平台,测序读长短,一般在150bp左右,即使采用双端测序技术,测得的有效读长也不超过500bp。而人类转录组中转录本的长度普遍在1000bp以上,有些转录本长度甚至超过100kb(Piovesan et al., 2016; Frankish et al., 2019),远远超过二代测序方法所能检测的最大读长。
为了解决基于二代测序平台的单细胞转录组测序技术难以获得单个细胞中全长转录本的准确信息这一核心困难, 2020年12月30日,北京大学未来基因诊断高精尖创新中心、生物医学前沿创新中心汤富酬课题组与北京希望组生物科技有限公司合作在Plos Biology上在线发表了题为“Single-cell RNA-seq analysis of mouse preimplantation embryos by third-generation sequencing”的研究论文。该研究的主要突破有:
1)开发了一种基于三代单分子测序平台的高灵敏度单细胞转录组测序方法—SCAN-seq (Single cell amplification and sequencing of full-length RNAs by Nanopore platform),能够在单细胞分辨率直接获取全长转录本序列信息,表现出高灵敏度和高稳健性,在小鼠胚胎干细胞每个单细胞中可以检测到8000多个基因的表达,与之前基于二代测序平台最灵敏的单细胞转录组测序方法不相上下(如图1所示)。
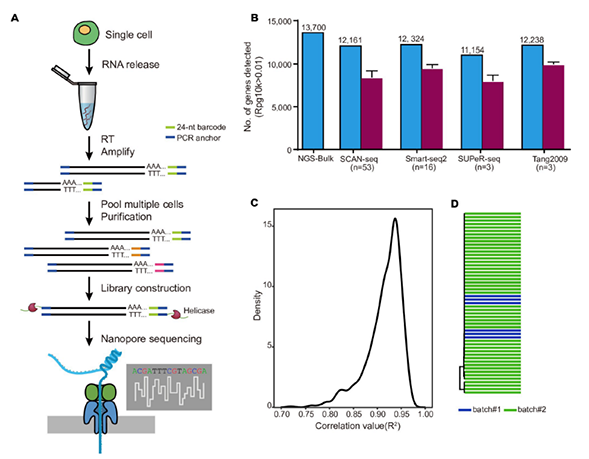
图1 SCAN-seq的流程和评估
2)鉴定出了30000多种全新的转录本。总共只测序了200多个单细胞就在小鼠胚胎干细胞(mESCs)和小鼠植入前胚胎中分别鉴定出6487条和27250种新转录本。相比基于二代测序平台的所有单细胞转录组测序方法,SCAN-seq能够区分新找到的转录本是来自同一已知转录本的新转录本,还是来自不同已知转录本已注释剪接点的重新组合的新转录本(如图2所示)。
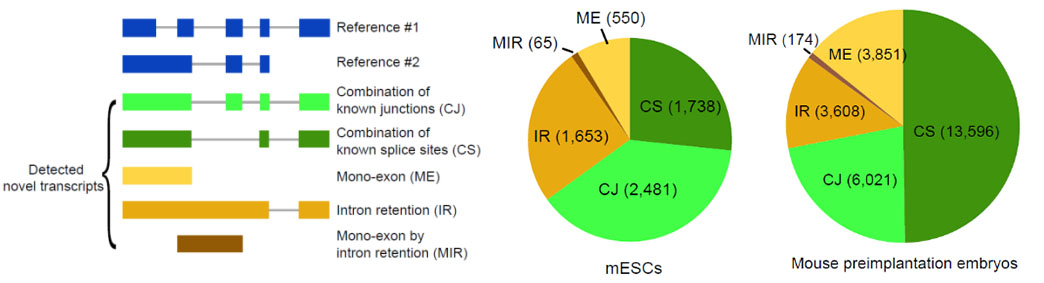
图2 未注释转录本的鉴定
3)首次提出单细胞转录组三代测序数据可以将一个单细胞中的父母源转录本准确区分开、分别进行精准定量分析。SCAN-seq显示出在同一个单细胞中精准识别小鼠品系特异性单核苷酸多态性(SNPs)的能力,平均误差率只有1.8%。利用这一方法,在单细胞分辨率确认了小鼠2-细胞期后的胚胎细胞中父源等位基因的mRNA比例逐渐增加,到囊胚期时每个胚胎细胞中来自母源和父源等位基因的mRNA拷贝数变得相当(如图3所示)。
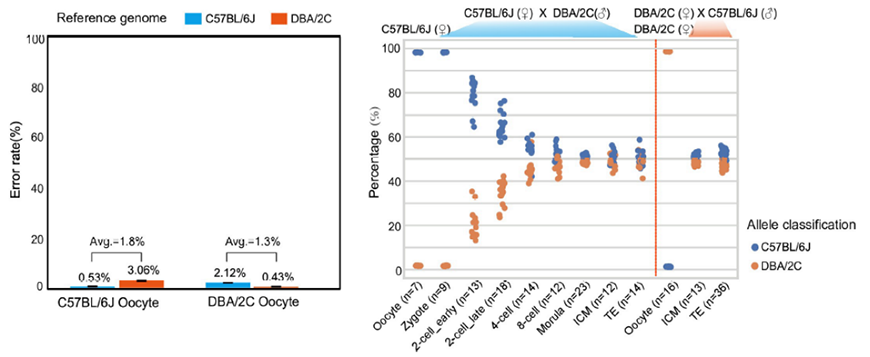
图3 等位基因特异性转录本的分析
该研究开发出的SCAN-seq新方法具有广阔的应用前景,能够克服单细胞转录组二代测序方法的各种局限性,将单细胞组学测序从“二”时代推进到“三”时代:(1)从一般只能测序单细胞中cDNA一端的有限信息,提升到能够测序单细胞中cDNA的全长信息;(2)从单细胞中一个基因的所有不同可变剪接产物(转录本)混合测量无法区分,提升到把单细胞中每个基因的所有不同可变剪接产物(转录本)精准分开;(3)从单细胞中一个基因的父母源表达信息混合在一起无法区分,提升到把单细胞中每个基因的父母源转录本精准分开;(4)从只能在单细胞中检测独特序列基因的转录本信息,提升到同时也能精准检测单细胞中高度重复序列的转录本信息;(5)从“一个基因,一个表型”的精度(one gene, one phenotype;人类基因组中有大约3万个基因),提升到“一种基因可变剪接转录本,一个表型”的精度(one RNA isoform, one phenotype;人类基因组中有大约30万种不同的可变剪接转录本)。总之,单细胞转录组三代单分子测序技术将揭开更多的转录组中“暗物质”的奥秘,给人类生物医学研究带来全新的发展机遇。
生物岛实验室研究员范小英、北京大学生命科学学院博士生廖雨涵和北京希望组生物科技有限公司汤冬硕士、李丕栋硕士为该论文的并列第一作者。北京大学未来基因诊断高精尖创新中心、生物医学前沿创新中心汤富酬教授与北京希望组王洋博士为该论文的共同通讯作者。该研究项目得到了国家自然科学基金委、北京市科技委和北京大学未来基因诊断高精尖创新中心的支持。
希望组作为三代测序的引领者,一直深耕三代测序领域,引进国际先进的PacBio Sequel II、ONT PromethION 48、MGISEQ2000、Bionano Saphyr光学图谱等技术平台,为科学研究和临床检测等提供多平台多水平的测序分析服务。利用单细胞结合三代测序平台,获取全长转录组信息,可为研究“一种基因可变剪接转录本,一个表型”打下夯实的基础。欲详细了解单细胞转录组三代测序服务及更多应用场景,可邮件联系sales-support@grandomics.com或联系希望组当地销售顾问。
参考文献:
- Tang, C. Barbacioru, Y. Wang, E. Nordman, C. Lee, N. Xu, X. Wang, J. Bodeau, B.B. Tuch, A. Siddiqui, et al. (2009). mRNA-Seq whole-transcriptome analysis of a single cell. Nature Methods, 6, 377-382.
Piovesan, A., Caracausi, M., Antonaros, F., Pelleri, M. C., & Vitale, L. (2016). GeneBase 1.1: A tool to summarise data from NCBI Gene datasets and its application to an update of human gene statistics. Database (Oxford), 2016, baw153.
Frankish, A., Diekhans, M., Ferreira, A. M., Johnson, R., Jungreis, I., Loveland, J., et al. (2019). GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 47, D766–D773.


 青岛星赛销售的单细胞拉曼分选–测序耦合系统(RACS-Seq)
青岛星赛销售的单细胞拉曼分选–测序耦合系统(RACS-Seq)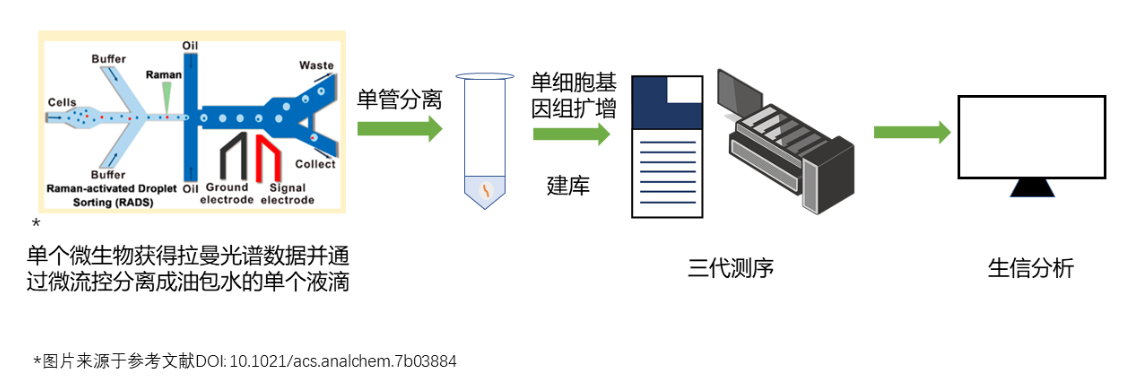

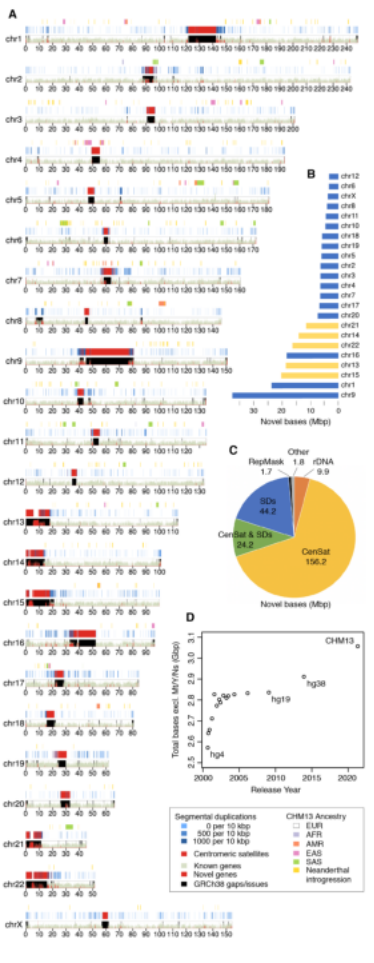


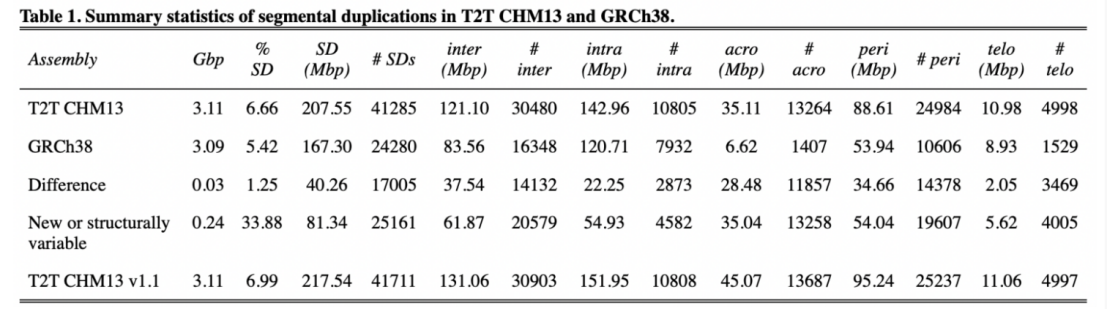
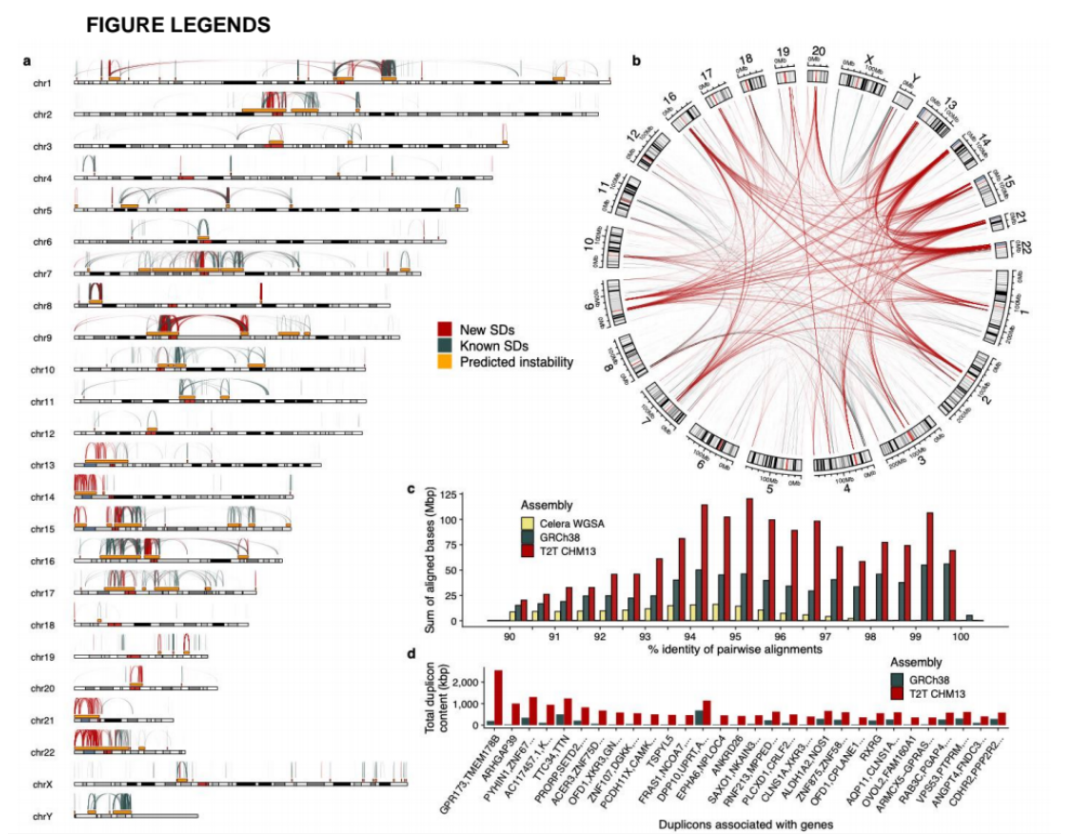

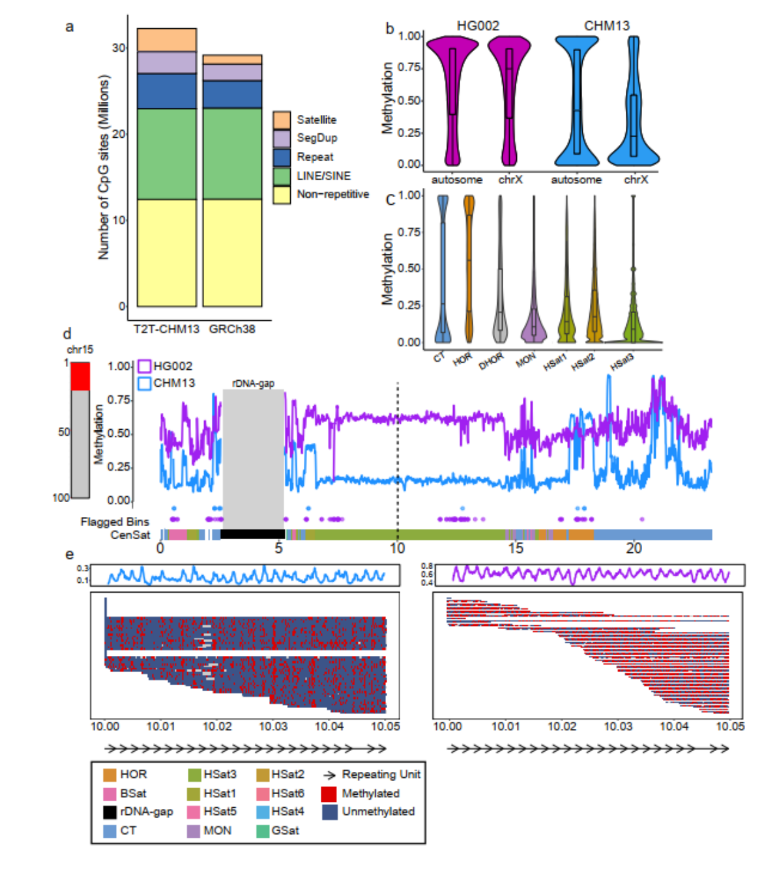




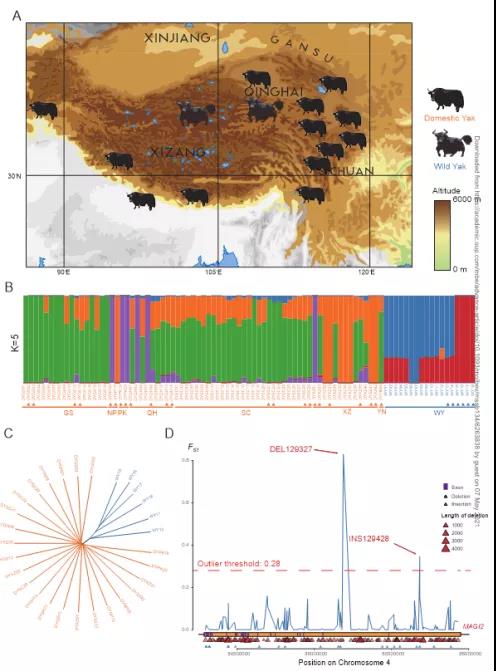
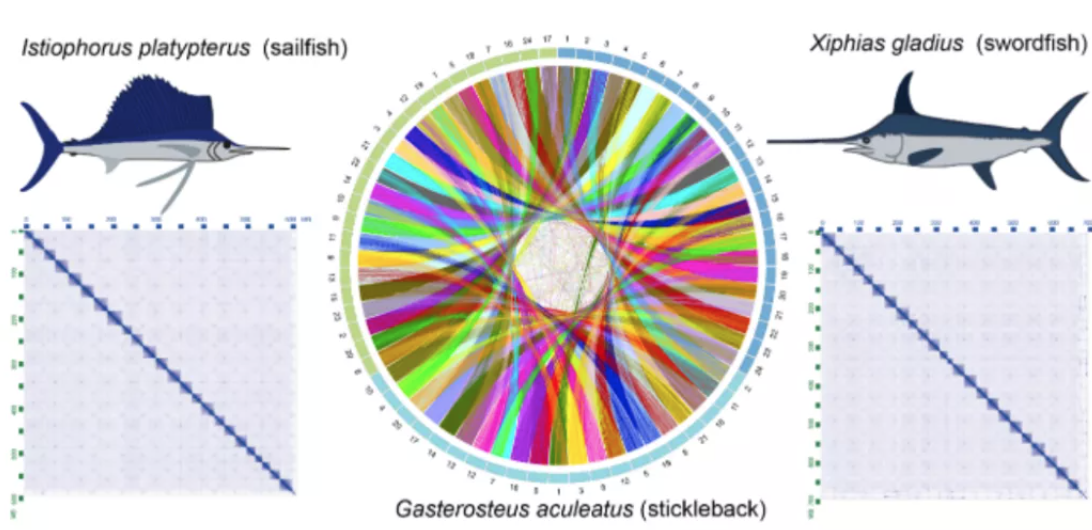
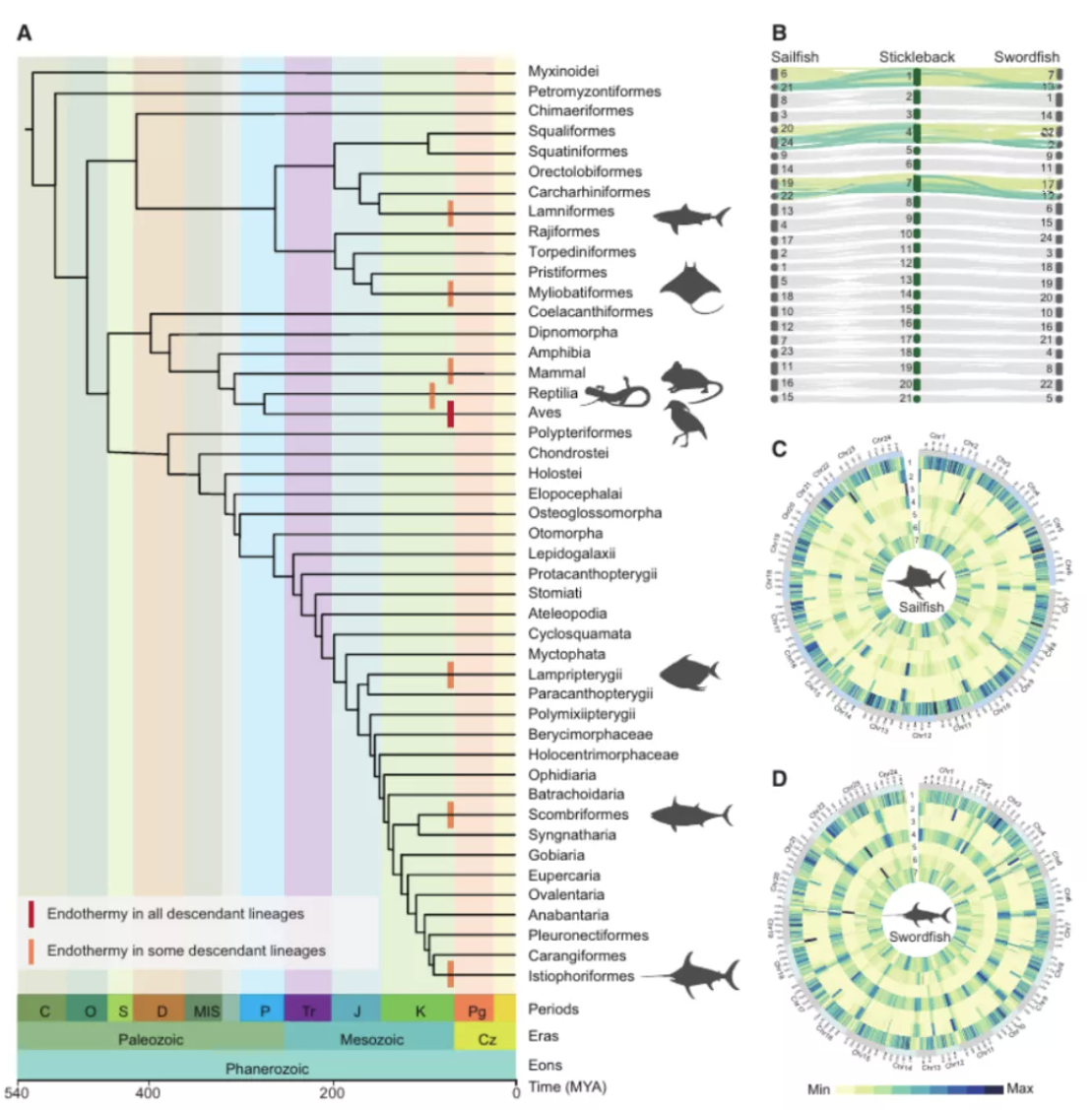
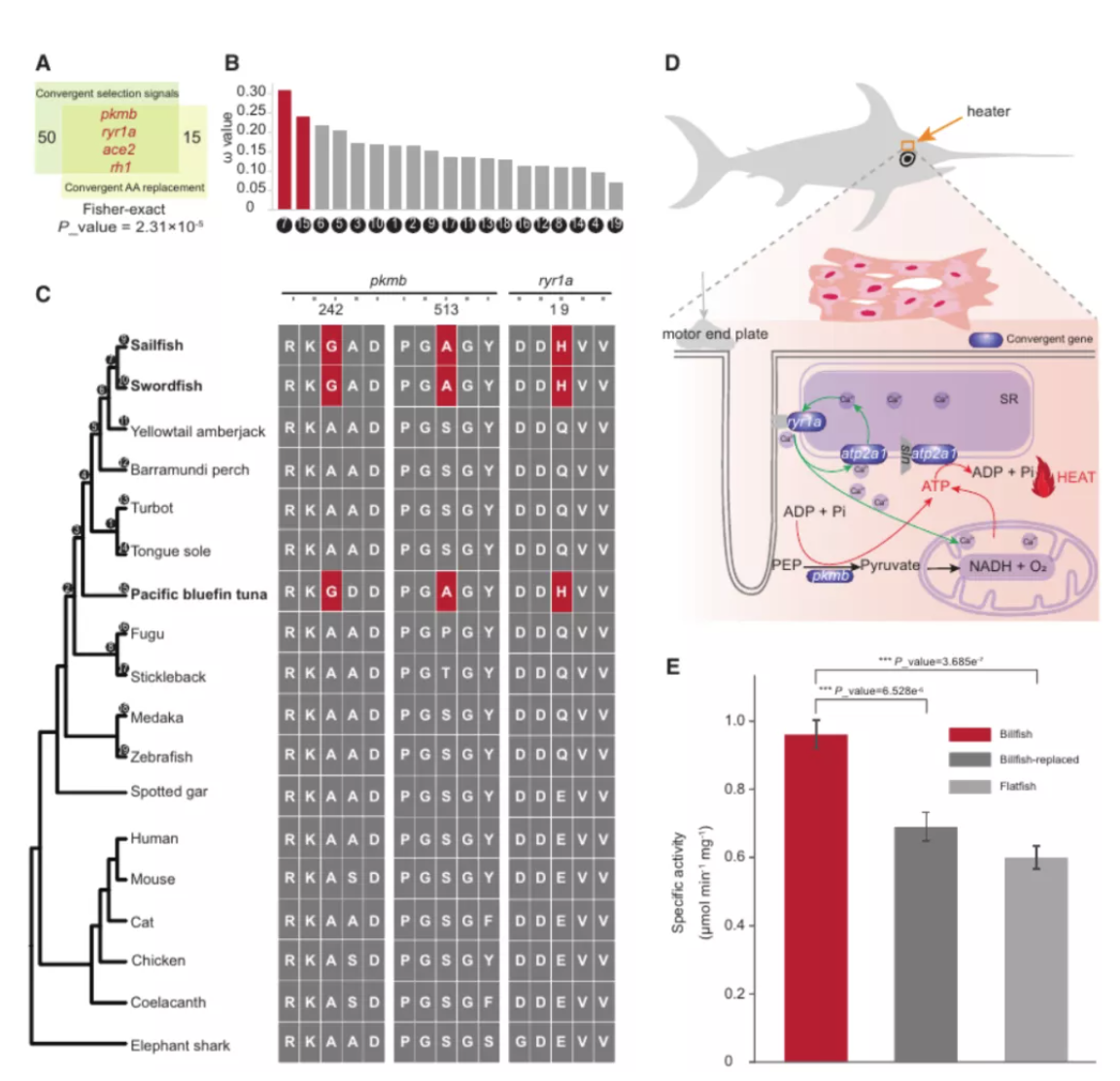

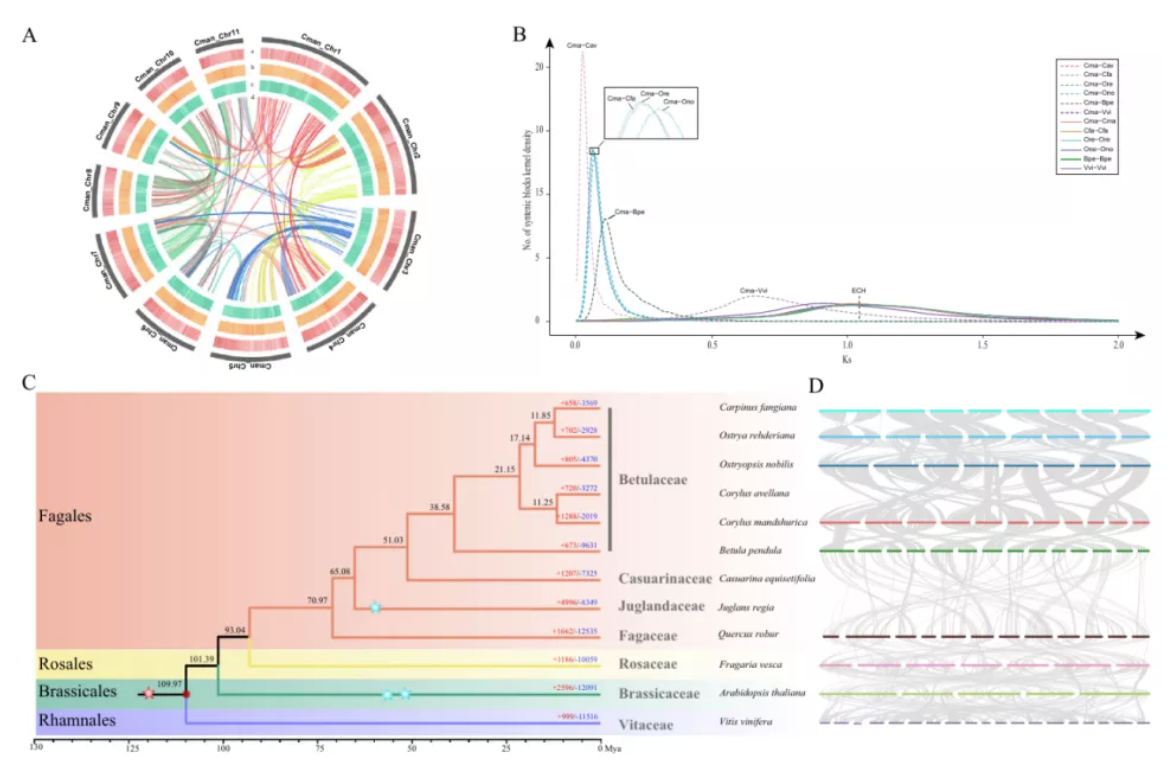
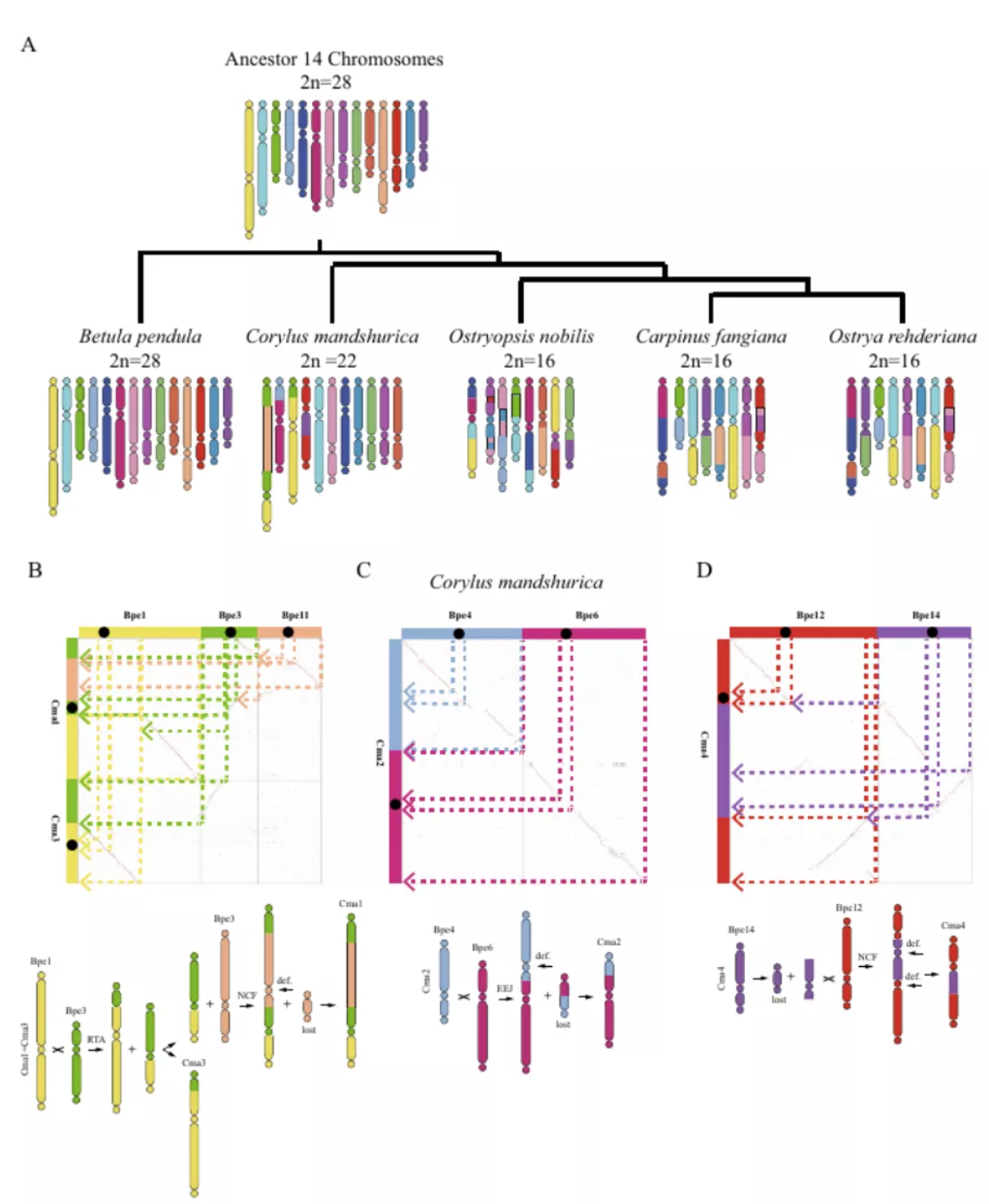
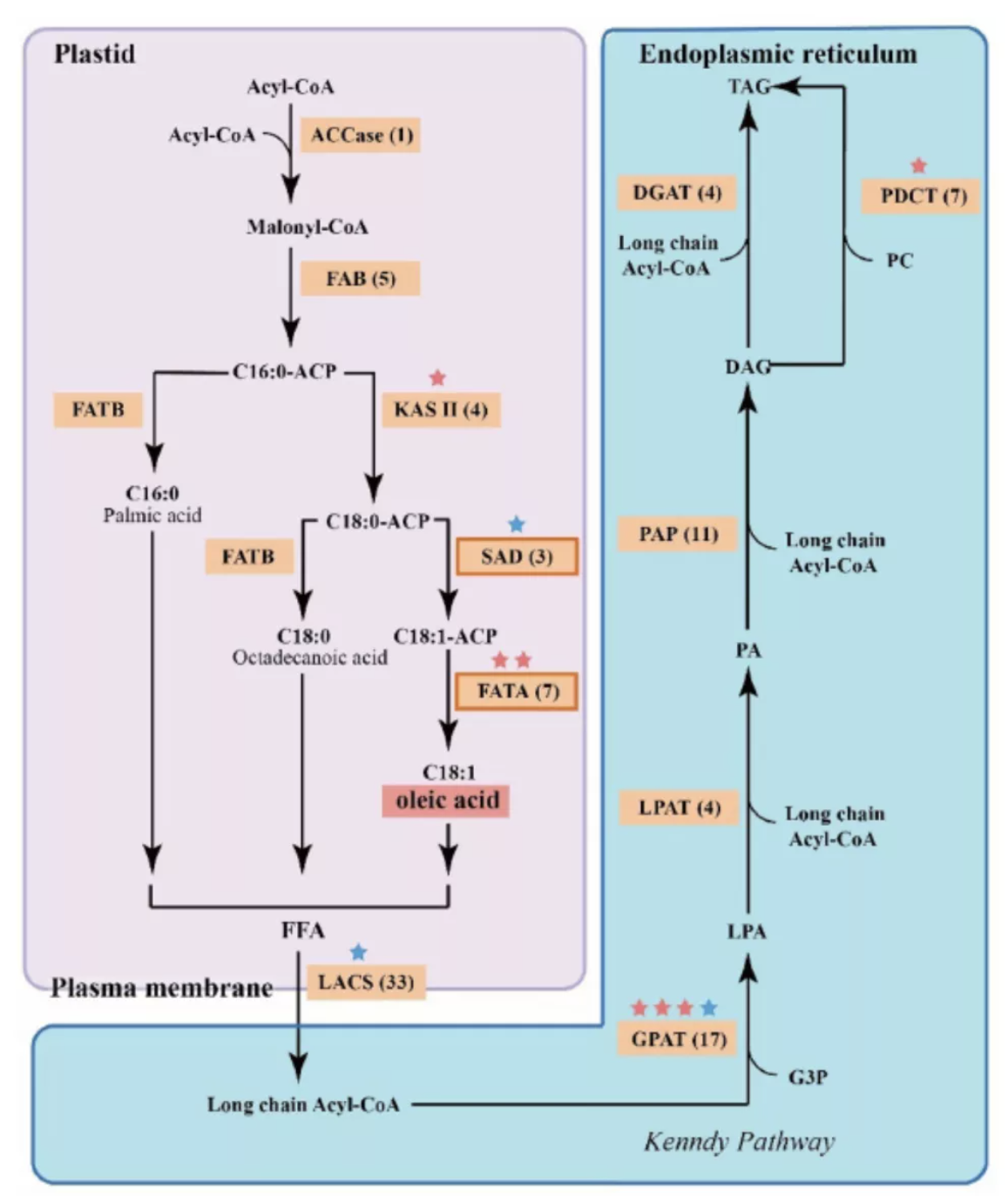

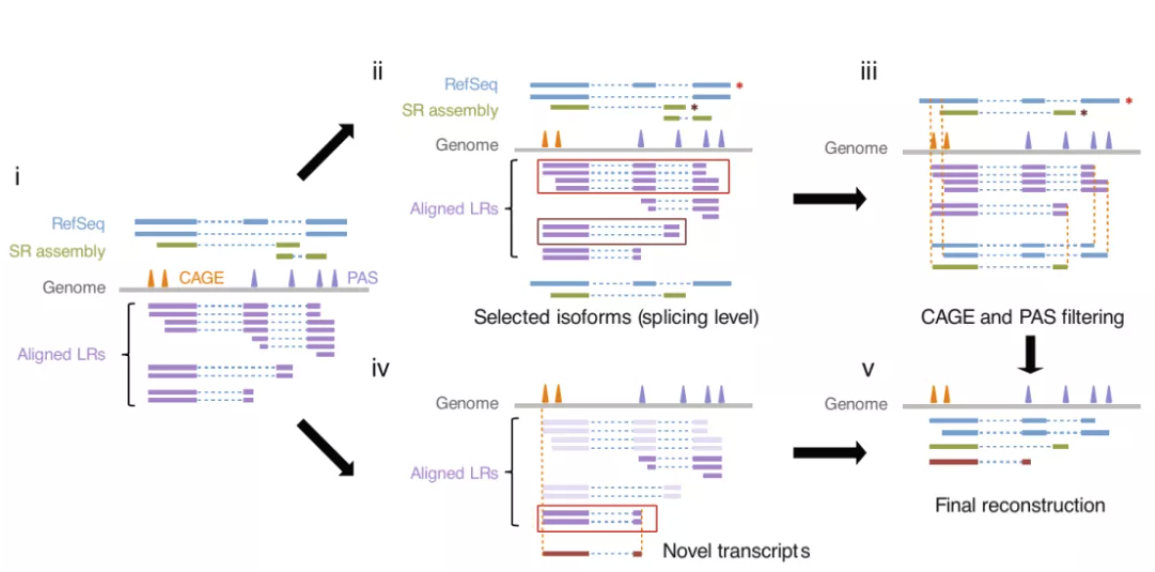
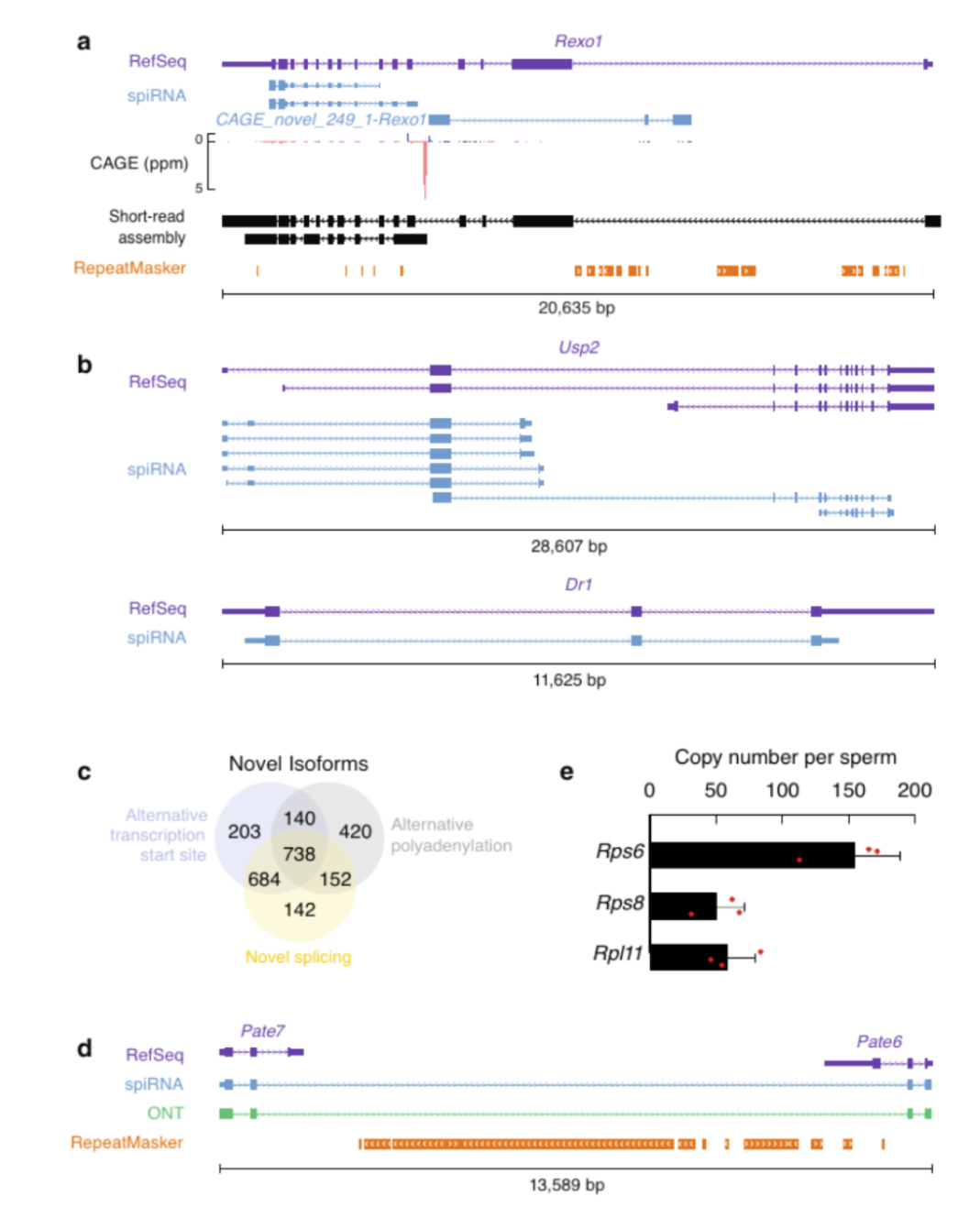
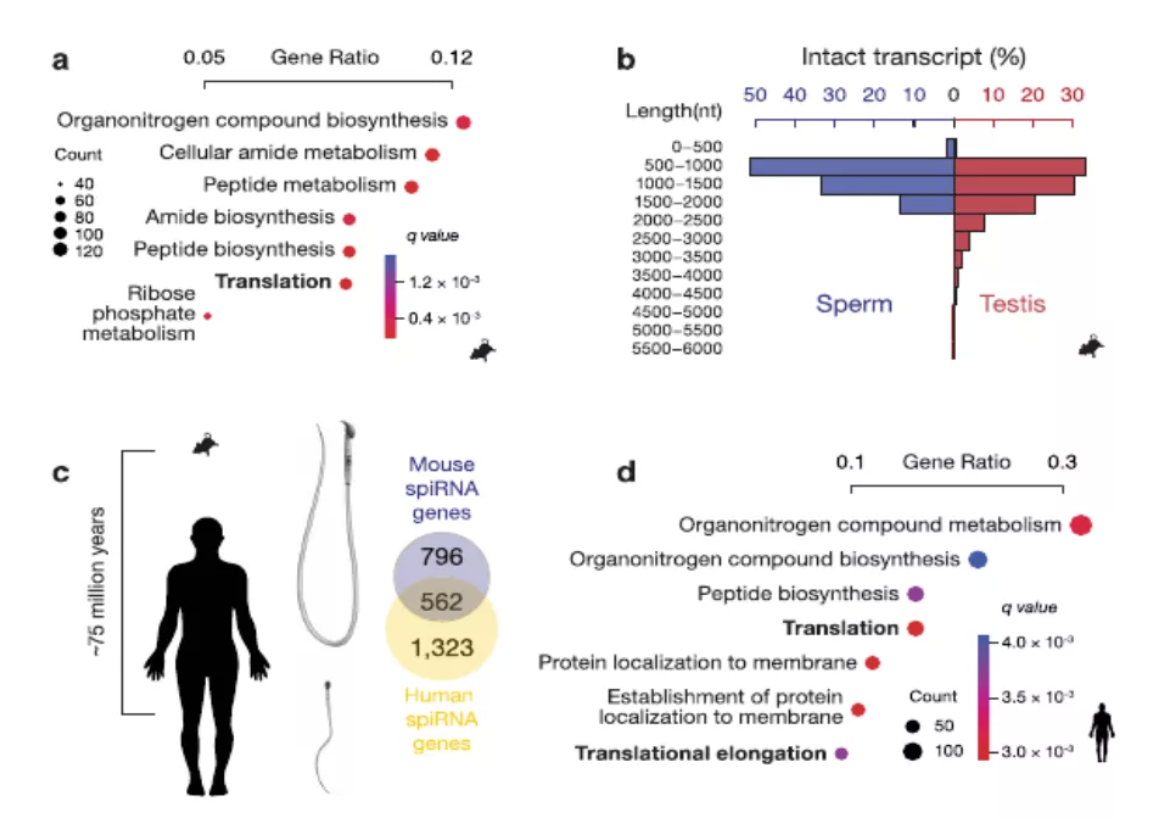
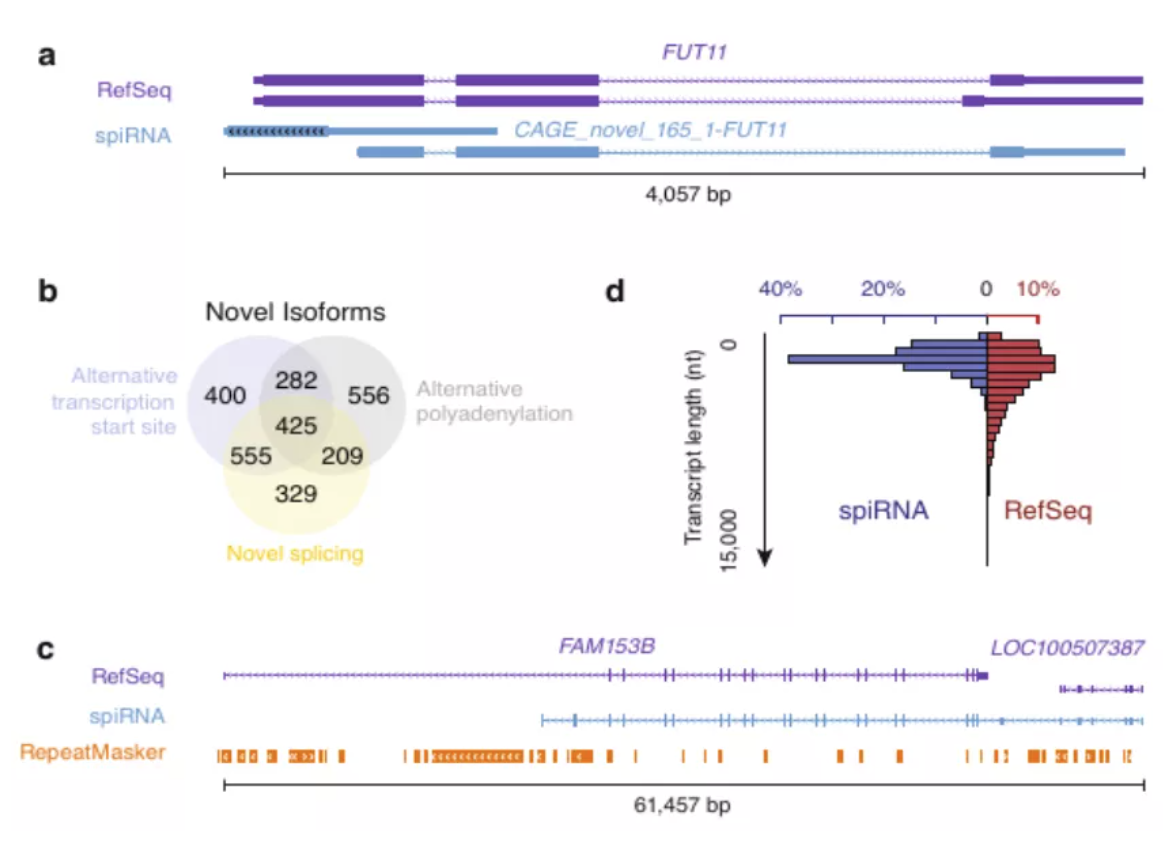
 图3 spiRNAs include both mRNAs and lncRNAs
图3 spiRNAs include both mRNAs and lncRNAs