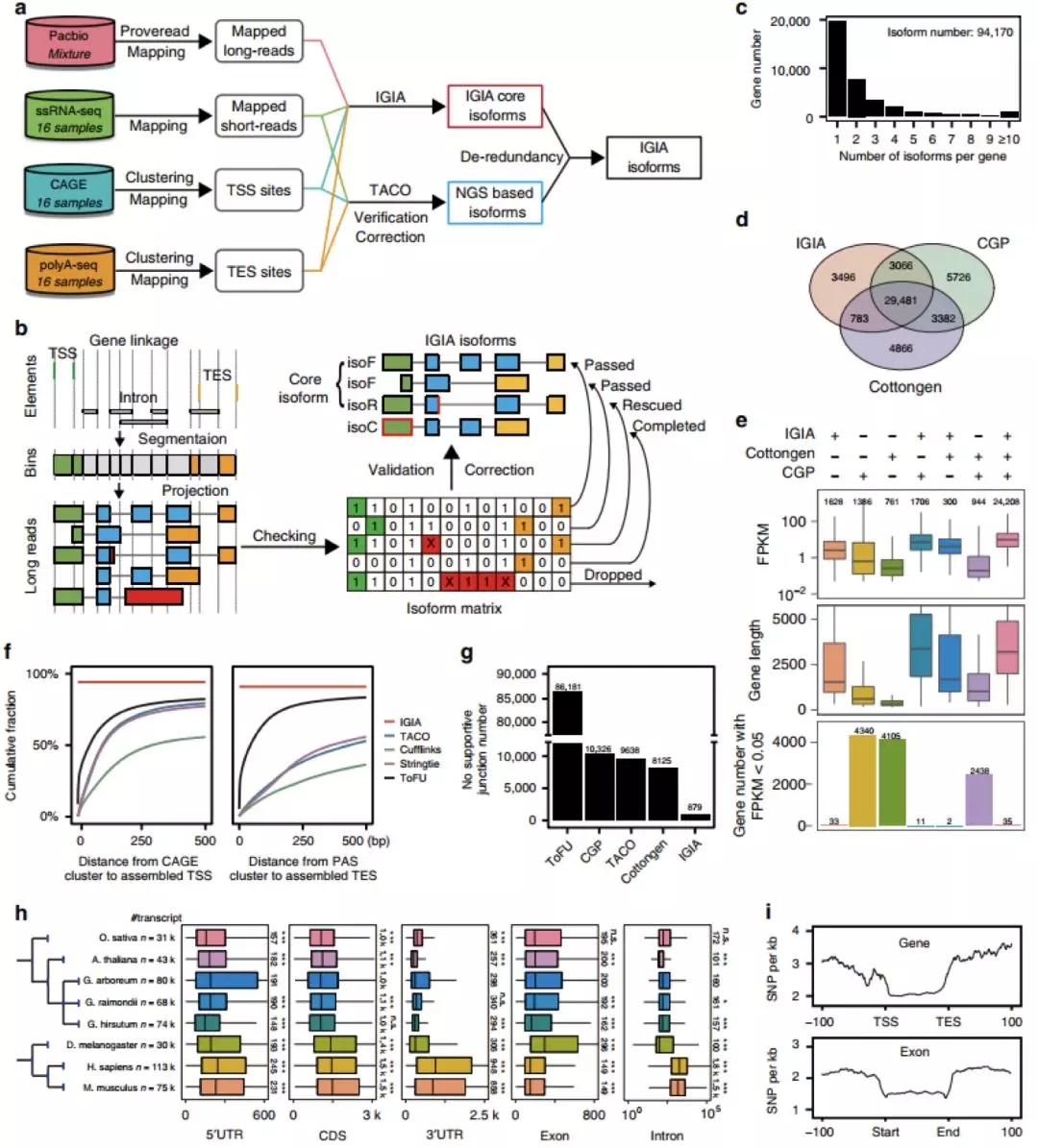
结构变异(Structural variants,SVs)通常是指基因组上大长度的序列变化和位置关系变化。研究表明,与单核苷酸多态性(SNPs)相比,SVs可以解释更多的表型变异。在植物基因组中,SVs的类型、大小以及对于表型的贡献多有报道,大概1/3已报道的作物表型是由于结构变异引起的(Gaut et al. 2018 Nature Plants),但是对于SVs在种群个体间的分布以及种群动态,人们知之甚少。希望组科技服务在6月份推出了基于Nanopore平台的三代测序群体基因组SVs研究,许多老师对这一研究非常感兴趣,但苦于没有研究思路。最近,Nature Plants杂志在线发表了一篇群体水平结构变异研究文章,加州大学Irvine分校周永锋博士为第一作者,Brandon Gaut教授(UC Irvine)和Dario Cantu教授(UC Davis)为共同通讯作者。该研究探讨了葡萄驯化过程中结构变异的群体遗传学,今天就给大家分享一下这篇文章的研究策略,给各位提供一些科研灵感。
研究背景
多年生植物栽培葡萄(Grapevine)是由其野生祖先欧亚葡萄(Eurasian grapevine),在约8000年前的高加索地区驯化而来。驯化提高了果实含糖量,增大了果实的体积和串大小,改变了种子形态,同时使雌雄异株转变为雌雄同体无性繁殖。无性繁殖作物处于永久性杂合状态,并随着时间累积体细胞突变(Zhou et al. 2017 PNAS)。理论上,雌雄同体葡萄可以自交,但实践中,其自交后代无法存活,可能是近亲繁殖暴露了杂合状态下的有害等位基因。因此,大多数葡萄品种是远源亲本之间的杂交种,加上体细胞突变的积累,导致葡萄品种往往是高度杂合的。本研究通过调查野生和驯化葡萄中SV的群体遗传来填补我们对植物基因组进化认知的空白。
研究策略

无性系繁殖葡萄基因组中肆虐的半合子状态
研究者首先利用三代测序+二代测序+Hi-C技术,组装了高杂合葡萄霞多丽品种的基因组序列,并对其进行了注释和评估,发现无性系繁殖葡萄基因组中有七分之一(~15%)的基因属于半合子,这一结果在黑比诺(PN40024)基因组与赤霞珠(Cab08)参考基因中得到了验证。
随后研究者用长、短reads比对和全基因组比对等方法,综合比较了Char04和Cab08两基因组之间的SVs。结果表明利用长reads比对检测到59,913个SVs,其中75%得到另外两种方法的证实。两个品种之间有近5%的PAV基因差异,半合子基因差异高达25%,表明葡萄品种之间显著的结构变异(图2)。

图2 高杂合Char04及与Cab08结构变异的比较
SVs群体遗传分析
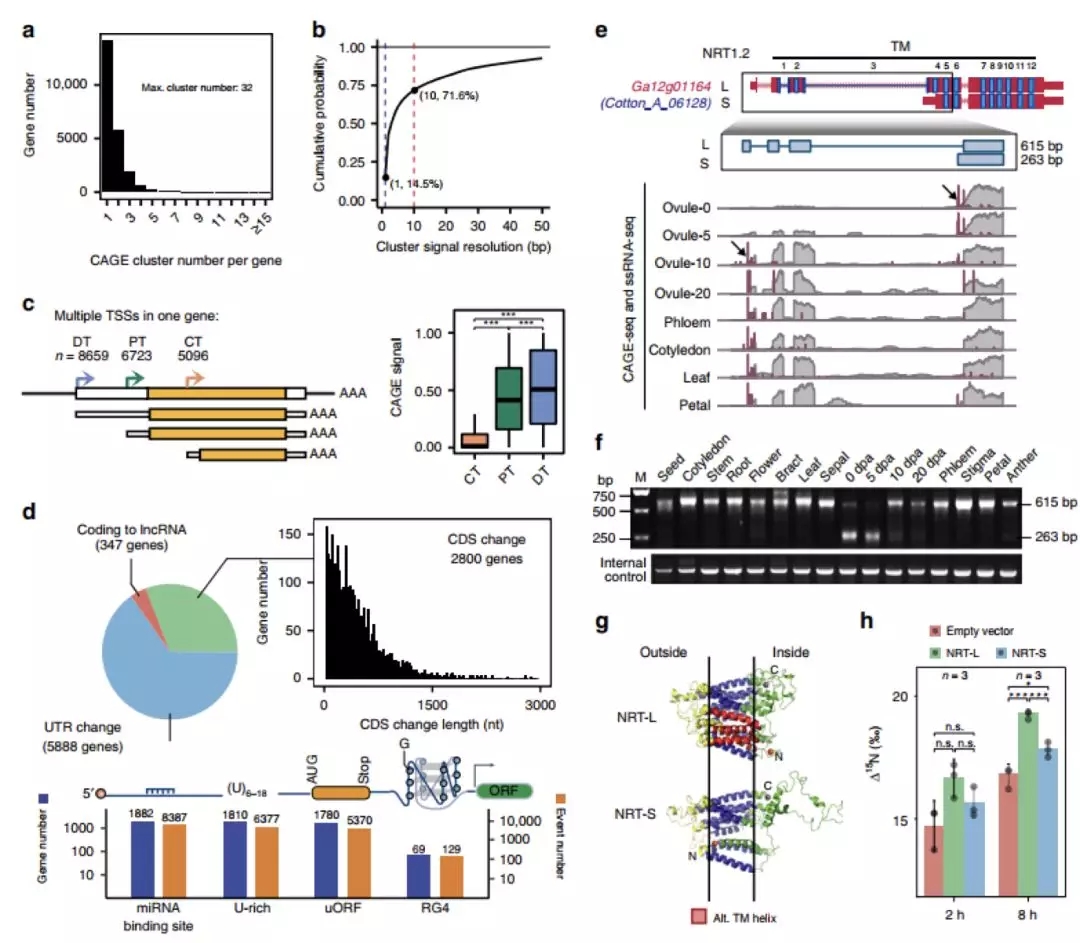
为了获得更广泛的葡萄品种及其野生亲缘SVs信息,研究者收集了有代表性的50个栽培葡萄品种和19个野生亲缘品种的短读长测序数据。以Char04为参考基因组,以Char04和Cab08综合比对的交叉SVs集合为金标准,获得了一组高度筛选的481,096个SVs。
随后,研究者利用上述SVs集合计算了12个野生种和12个栽培种的SFS(图3),推断了对SVs类型的选择强度,并对比了驯化和野生祖先之间的SVs频率。结果非同义SNP(nSNP)和SVs都经历了强烈的纯化选择,不同SVs类型中,易位TRAs和倒位INVs的选择性更强。因此SVs事件比nSNP更有害,INV和TRA事件尤其有害。

图3 处于强烈净化选择中的有害SVs
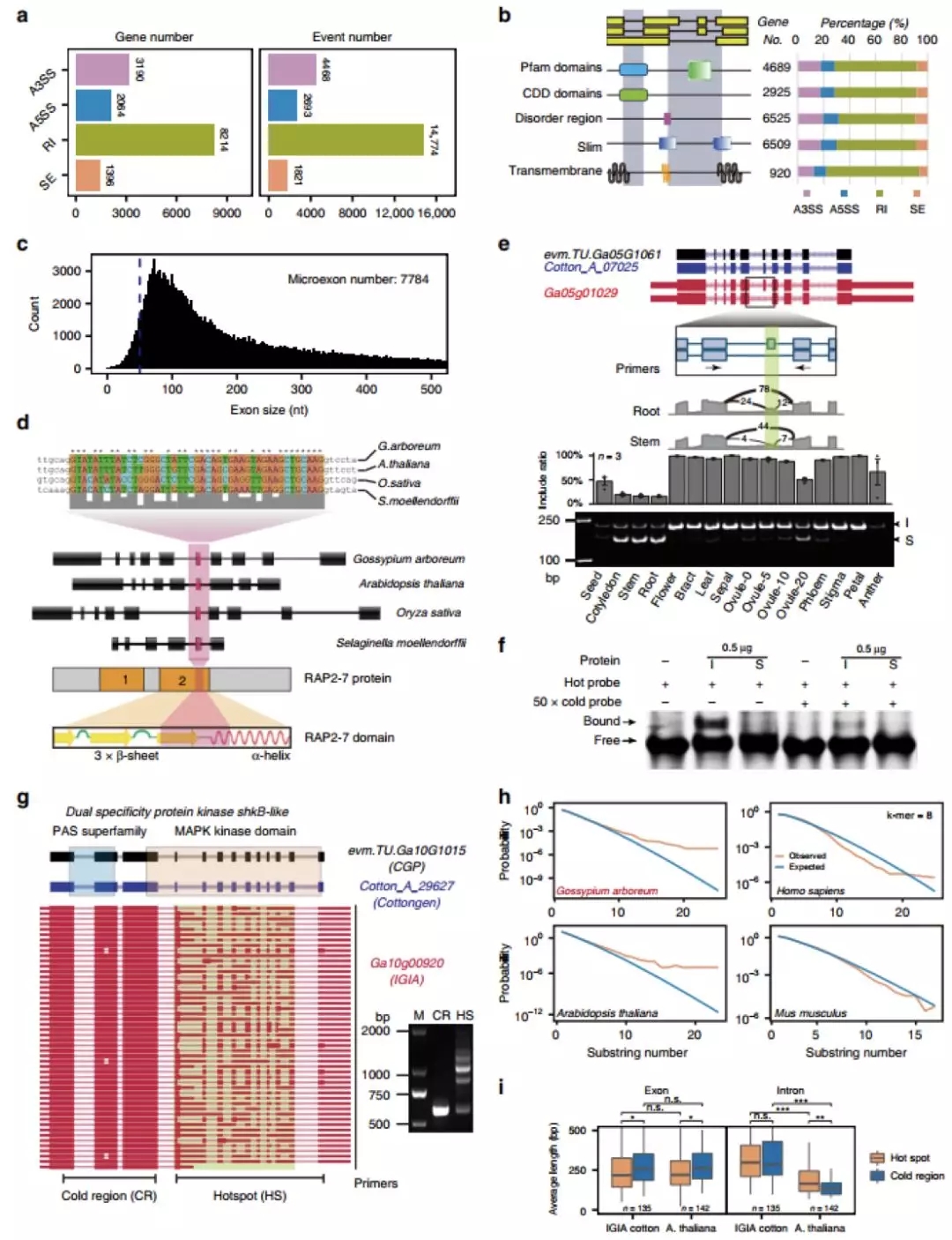
SVs在无性系繁殖体中积累
研基于SNP的个体杂合度分析,栽培葡萄个体杂合度要高出野生型葡萄11%,相应的单个栽培种SVs比野生种高出6%,但纯合子SVs或推测为中性的sSNPs没有明显增加(图4a)。因此有害SVs在无性繁殖情况下以杂合隐性形式隐藏、积累。
杂合变异的积累会影响连锁不平衡(LD),通过测量SVs、SNP和组合数据集的LD随物理距离的下降来分析SVs的种群频率。结果发现,与野生品种相比,栽培品种的LD下降速度更快;与SNP相比,SVs的LD下降更快;下降速度最快的是SV+SNP数据集。表明由于有害影响,SVs通常比SNP的种群频率更低。


图4 葡萄驯化相关SVs的群体遗传学
大的,独立的倒位对浆果颜色的影响
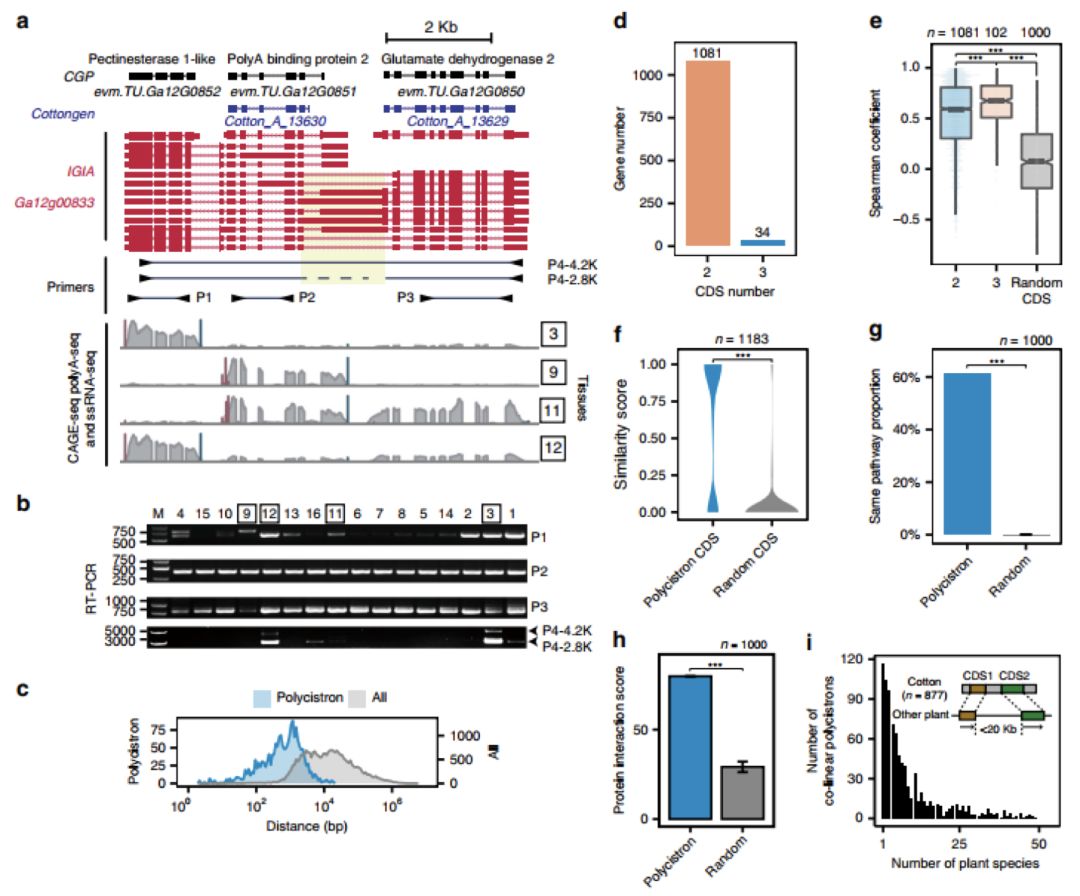
通过计算固定指数(FST)来估计SNP和SVs在基因组中的差异(图4c),在2号染色体上发现了两个异常值分别与性别决定和浆果颜色相关。其中对浆果颜色相关区域的进一步研究发现,在霞多丽中有一个4.82Mb的倒位(图5b),并有证据支持白浆果的独立起源通常是由这种倒位介导的(图5d),其导致了半合子状态的花青素合成基因MybA1和MybA2的等位基因空缺。

图5 与白色浆果相关的染色体倒位
小结
本研究首先组装了高杂合葡萄霞多丽的基因组序列,评估了该基因组中SVs类型和分布以及导致遗传半合子的SVs。随后将霞多丽与赤霞珠基因组进行综合比较,获得了一套种间SVs标准集,并以此指导、推断栽培葡萄及其野生祖先群体样本中的SVs。然后利用获得的群体SVs数据集,推断不同类型变异的选择强度,探讨了在栽培葡萄上由异交向无性繁殖转变的效应,最后研究了栽培葡萄与其野生祖先之间SVs差异特别显著的与浆果颜色相关的基因区域。 在结构变异的研究中,最首要的任务是获得到研究对象全面、准确的SVs集合,本研究中作者为了获取准确的SVs集合,利用三代测序组装了霞多丽基因组,采用长读长比对来鉴定SVs,基因组比对和短读长比对进行验证,短读长仅检测到长读长比对检测数量的62%,长读长检测的SVs中75%得到另外两种方法的验证。可见相比短读长利用三代测序检测的SVs更加全面准确。
参考文献:
Gaut B S, Seymour D K, Liu Q, et al. Demography and its effects on genomic variation in crop domestication[J]. Nature plants, 2018, 4(8): 512.
Zhou Y, Massonnet M, Sanjak J S, et al. Evolutionary genomics of grape (Vitis vinifera ssp. vinifera) domestication[J]. Proceedings of the National Academy of Sciences, 2017, 114(44): 11715-11720.