萤火虫是鞘翅目萤科的昆虫,成虫个体一般较小,大多数体长1厘米,少数种类可以达到3厘米。雌性个体要略微大于雄性个体。萤火虫最独特的特征是腹部具有特化的发光器,不同萤火虫之间发光器区别很大,这也是萤火虫分类的重要特征之一。世界上已知萤火虫有2000多种,全世界仅发现了8种水栖萤火虫,原先都隶属于萤科第一大属——熠萤属。付新华教授与合作者以武汉萤为模式种,确立了一个新属——水萤属。
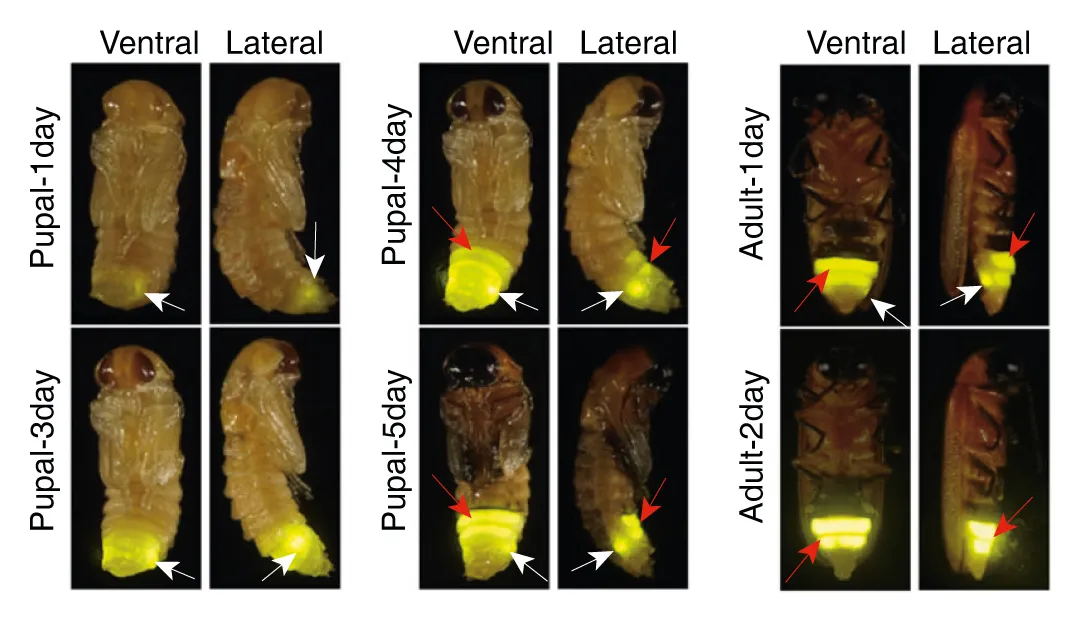
图1 雷氏萤A. leii的发育阶段。幼虫期1天至成虫期2天,白色箭头表示幼虫的发光器,红色箭头表示成虫的发光器
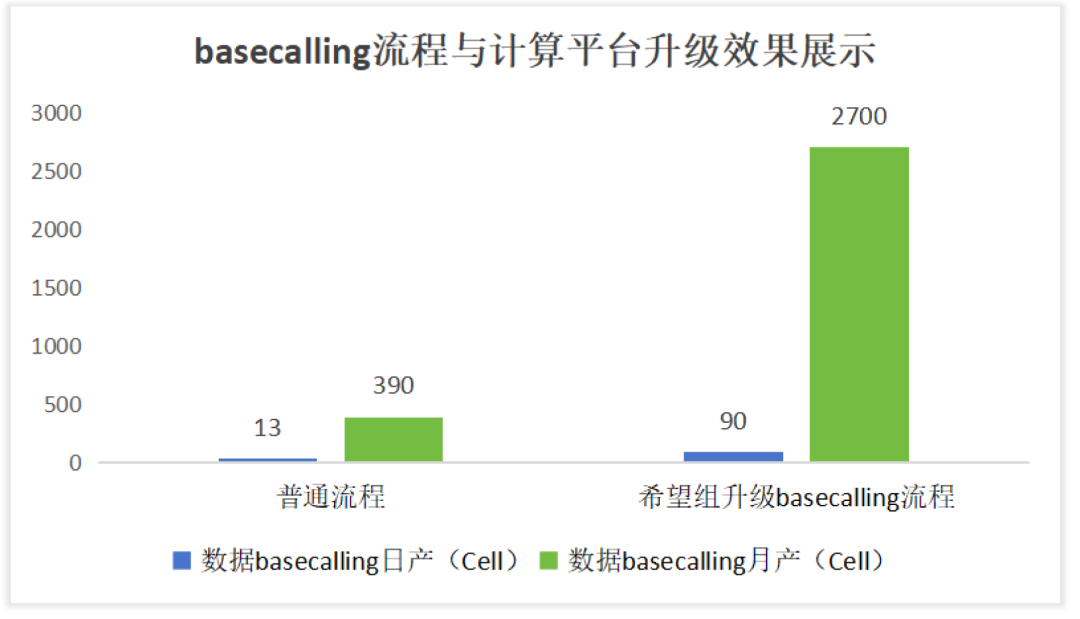
希望组与华中农业大学植物科学技术学院付新华教授的首次合作在2017年,基于PacBio Sequel(~73X)平台测序,完成了首个萤科物种胸窗萤(Pyrocoelia pectoralis)的高质量基因组测序组装,发表了《Long-read sequence assembly of the firefly Pyrocoelia pectoralis genome》文章[未来组项目文章] 三代萤火虫基因组文章发表。随着测序技术的不断升级,希望组与付新华教授再次合作,通过单分子纳米孔测序(ONT,~170X)与Hi-C挂载组装了染色体水平的水栖萤火虫雷氏萤(Aquatica leii)基因组。付新华和朱馨蕾依托高质量的雷氏萤基因组,结合不同发育阶段的蛹的比较转录组学,成功筛选出调节A. leii 成虫发光器发育的两个关键转录因子AlABD-B和AlUNC-4,于2024年3月5日在《Nature Communications》上发表题为《Key homeobox transcription factors regulate the development of the firefly’s adult light organ and bioluminescence》的研究成果!

这篇论文研究了水栖萤火虫成虫发光器发育和生物发光的分子调控机制。通过单分子纳米孔测序(ONT)和高通量染色体构象捕捉测序技术组装了水栖萤火虫雷氏萤(Aquatica leii)的染色体水平基因组,并发现了两个关键的Homeobox转录因子AlABD-B和AlUNC-4调节着萤火虫成虫发光器的发育和生物发光。干扰这两个关键基因的表达会导致成虫发光器发育不完整或不发光。此外,AlAbd-B 和 AlUnc-4 激活了 AlLuc1 基因的表达以及一些过氧化物酶跨膜转运蛋白的表达。四个过氧化物酶转运蛋白参与将荧光素酶转运到过氧化物体内。这项研究为了解萤火虫成虫发光器的发育和生物发光提供了重要的研究基础。希望组为本项研究提供ONT等基因组测序、基因组组装、Hi-C挂载、注释和比较基因组分析等服务。
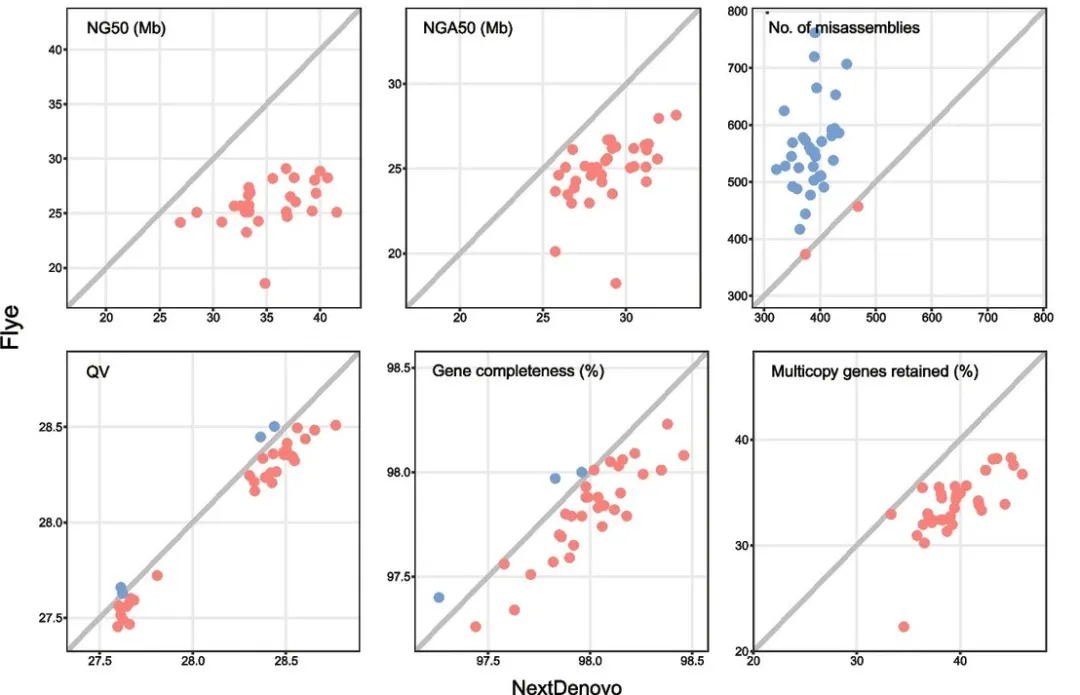
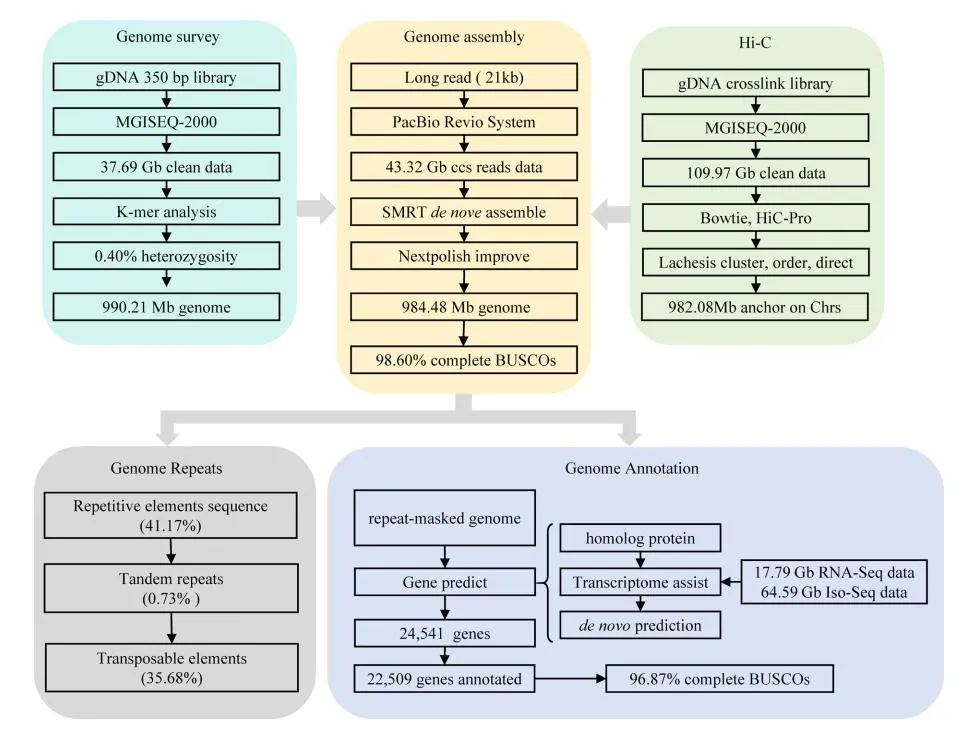
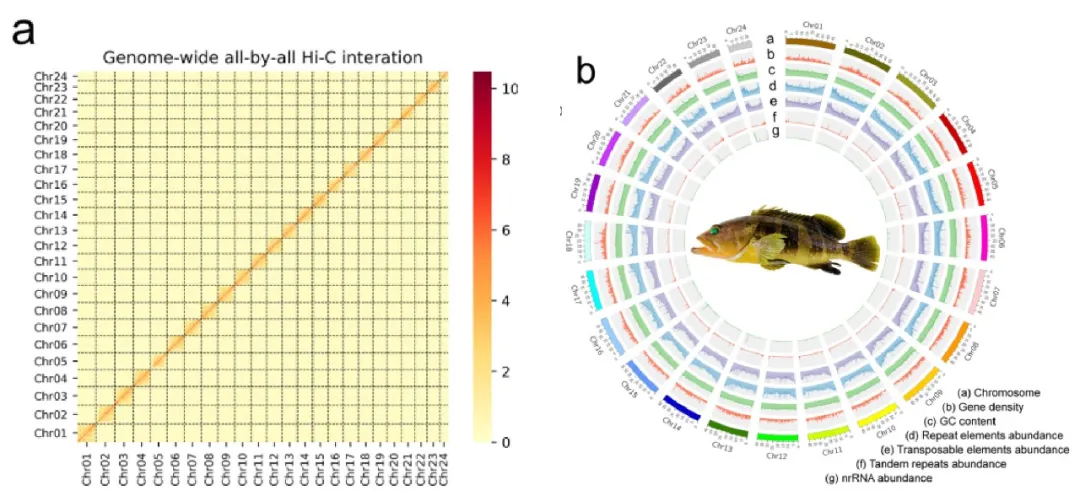
雷氏萤(Aquatica leii)组装基因组大小为1.04G,杂合度3.2%,包含222条contig,其中scaffold 和 contig 的 N50 值分别为 125.64 Mb 和 10.81 Mb。此研究分析了染色体数目和核型(2n = 14 + XY),并对基因组进行 Hi-C 挂载,挂载出 8 条染色体,同时统计了基因组的GC 含量、重复密度、 基因密度、转录因子密度、Homeobox 家族基因(图2D)。与胸窗萤(Pyrocoelia pectoralis)的高质量基因组(基因组大小760.4Mb,contig N50为3.04Mb,杂合度2%~3%)相比,基因组组装质量有了显著提升!
结合转录组数据,对雷氏萤(A. leii)进行基因组注释,共鉴定出16,472个基因。其中,14,874个基因(占90.30%)在至少一个公共数据库(Swissprot、NR、KEGG、GO和KOG)中进行了功能注释。为了推断 A. leii 的进化历史和系统发育关系,该研究使用了四个已发表的其他萤火虫基因组和六个鞘翅目昆虫基因组以及一个果蝇基因组作为外群,使用 OrthoMCL 进行了基因家族聚类分析。在这十二种物种中鉴定出了1,633个单拷贝同源基因用于构建系统发生树(图2E)。系统发生推断表明,A. leii和边褐端黑萤Abscondita terminalis是姐妹群,同属于熠萤亚科,根据 mcmctree 计算的分歧时间表明,A. leii在大约57.38百万年前从该亚科的其他成员的共同祖先中分离出来(图2E)。
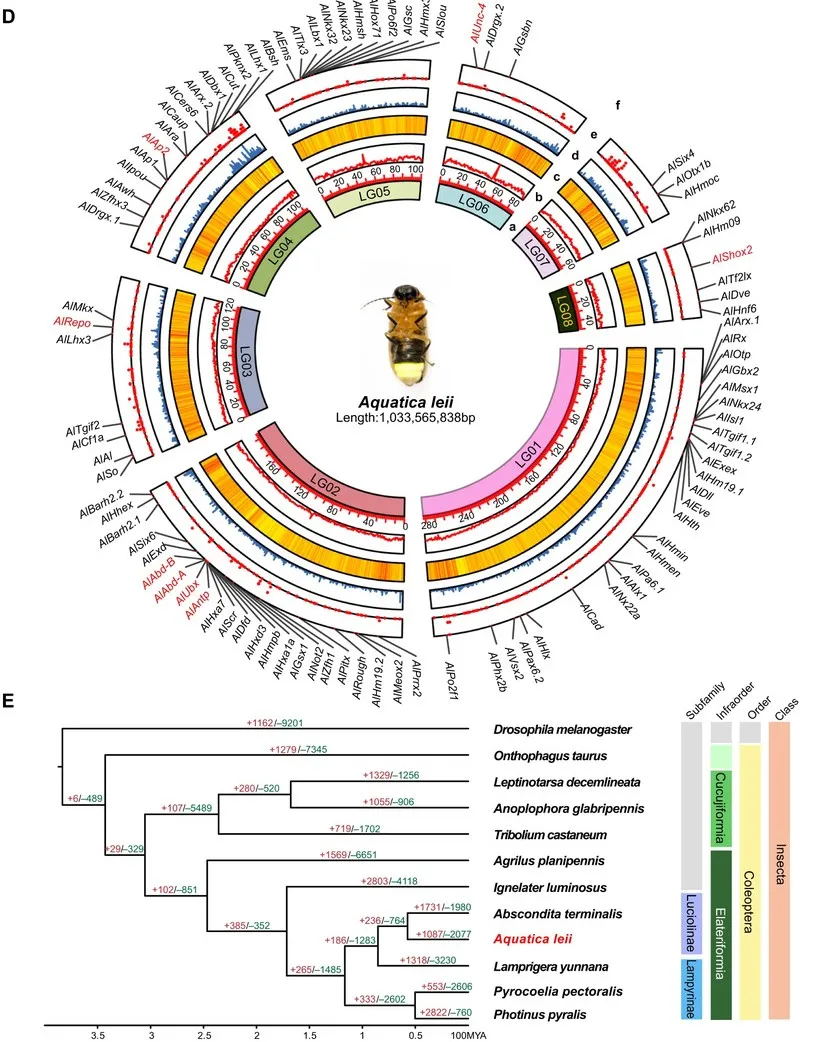
图2 雷氏萤A. leii 基因组特征圈图。红色标记基因在后续实验中被验证功能。
该研究使用AnimalTFDB(v3.0) 数据库比较了所有的 A. leii 蛋白,并获得了相应的A. leii 转录因子家族。在 A. leii 基因组中鉴定出了 914 个转录因子。根据功能域特征,将转录因子超家族成员分为 45 个不同的类型。其中,Homeobox 转录因子类型是A. leii 中除“锌指”转录因子类型之外最常见的转录因子类型,并且Homeobox 基因家族中的大量基因编码了在动物胚胎发育中细胞分化和发育中起关键作用的DNA结合同源域。结合转录组数据来分析Homeobox基因的表达模式,发现在蛹化过程中,只有六种homeobox基因(AlAbd-B、AlAntp、AlUnc-4、AlShox2、AlRepo和AlAp2)持续上调(p <0.05),而其他基因下调或其调控从上调变为下调。运用基因敲除技术研究发现,AlAbd-B 和 AlUnc-4 的敲除导致了不发光和过氧化物酶体空腔,这两个基因可能是萤火虫成虫发光器发育所必需的关键调节因子(图3)。
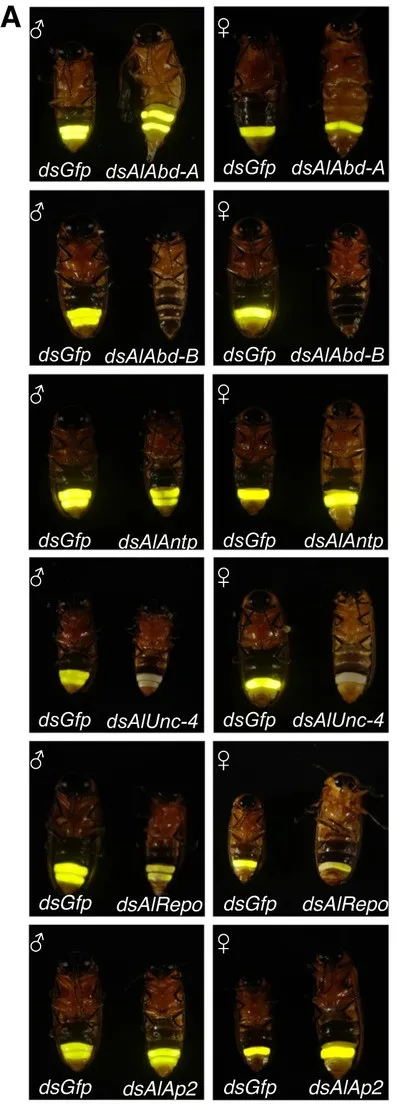
图3. A. leii 中与发光器发育和发光相关的6个homeobox基因的功能验证
萤火虫成虫发光器发育和发光存在两个关键步骤:一个是荧光素酶基因的表达,另一个是荧光素酶被转运到发光器,即生物发光发生的细胞器。该研究假设干扰荧光素酶基因表达或荧光素酶运输导致过氧化物酶体空腔。为验证这一假设,该研究对在蛹期敲除 AlAbd-B 和 AlUnc-4 的 1 日龄雄萤的转录组与对照组(Gfp 注射)进行了比较。结果表明,在 dsAlAbd-B 和 dsAlUnc-4 敲除组中,AlLuc1 的表达显著降低(图3A)。转录组测序分析和实时荧光定量PCR验证表明:(1)AlAbd-B 和 AlUnc-4基因的干扰,导致 AlLuc1、AlPx11c.2、AlPex5 和 AlPxmp2 的表达水平显著降低;(2) AlAbd-B 基因的敲除,导致AlPex1、AlPex13、AlPex14 和 AlPex16 的表达水平显著降低;(3)有趣的是,AlAbd-B 基因的敲除导致AlUnc-4 的表达水平显著降低。在这些基因中,该研究选择了AlLuc1 和其同源基因 AlLuc2 以及七个过氧化物酶基因AlPx11c.2、AlPxmp2、AlPex5、AlPex13、AlPex14、AlPex16、和 AlPex1 进行详细的功能研究(图4)。
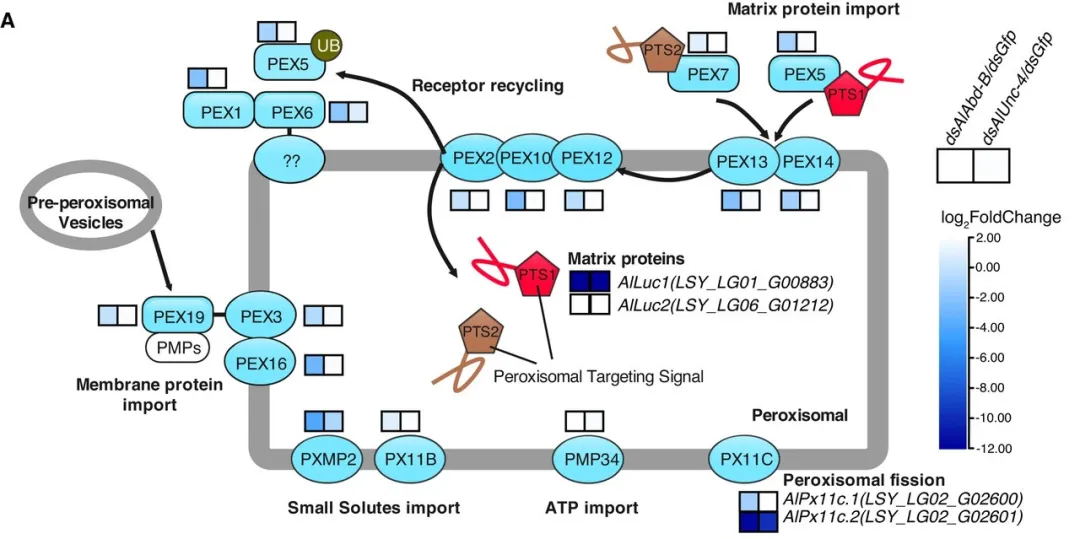
图4 AlAbd-B RNAi和AlUnc-4 RNAi表型与对照组相比转录组的差异基因表达分析
在AlLuc2敲除后,只有蛹的发光器发出荧光,表明成虫发光器的发育与 AlLuc2无关,因此该研究重点关注了 AlLuc1。dsAlAbd-B 和 dsAlUnc-4 组的转录组中,AlLuc1 的表达水平显著降低。通过 JASPAR 数据库分析,识别出每个转录因子的两个潜在 DNA 结合域。该研究假设 AlABD-B 和 AlUNC-4 与 AlLuc1 启动子相互作用并上调其活性,并进行了酵母单杂交 (Y1H)、电泳迁移率shift 实验(EMSA)、双荧光素酶报告基因检测、Western 蛋白印迹和免疫荧光 (IF) 检测来验证这一假设。最终揭示AlABD-B和AlUNC-4是发光器中AlLUC1表达所必需的(图5)。
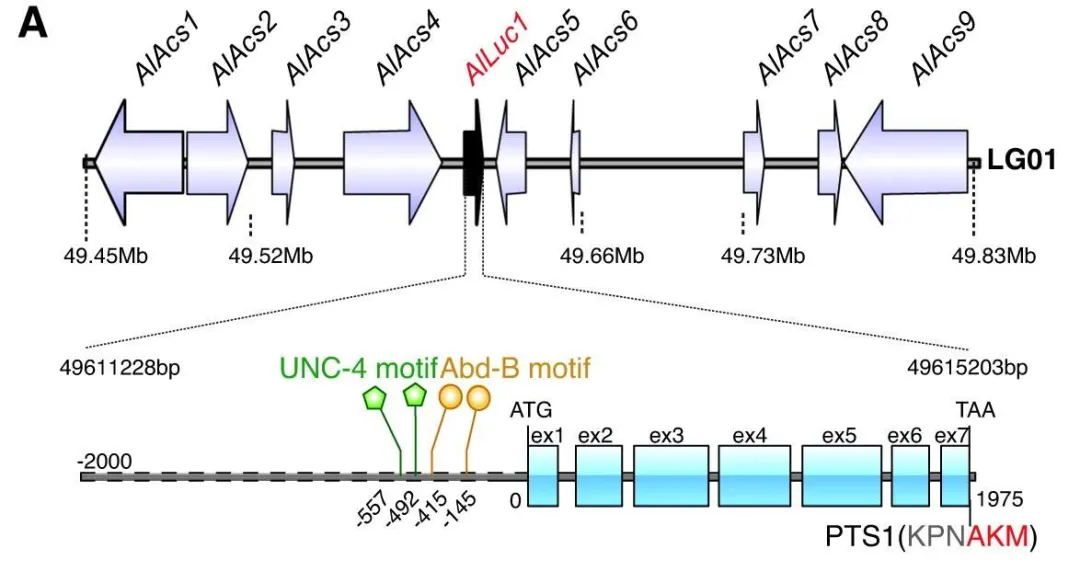
图5 Alluc1的基因组位点(顶部)和包含七个外显子的基因结构(底部)
萤火虫的荧光素酶AlLUC1在过氧化物酶体中起作用,这表明AlLUC1需要某些过氧化物酶体运转蛋白才能进入过氧化物酶体。该研究进行了RNAi分析,以验证筛选出的过氧化物酶(AlPX11C.2、AlPXMP2、AlPEX5、AlPEX13、AlPEX14、AlPEX16 和 AlPEX1)是否参与了ALLUC1 的导入。结果表明,AlPEX13、AlPEX14、AlPEX5和AlPXMP2蛋白参与了A.leii 中AlLUC1进入过氧化物酶体的过程(图6)。
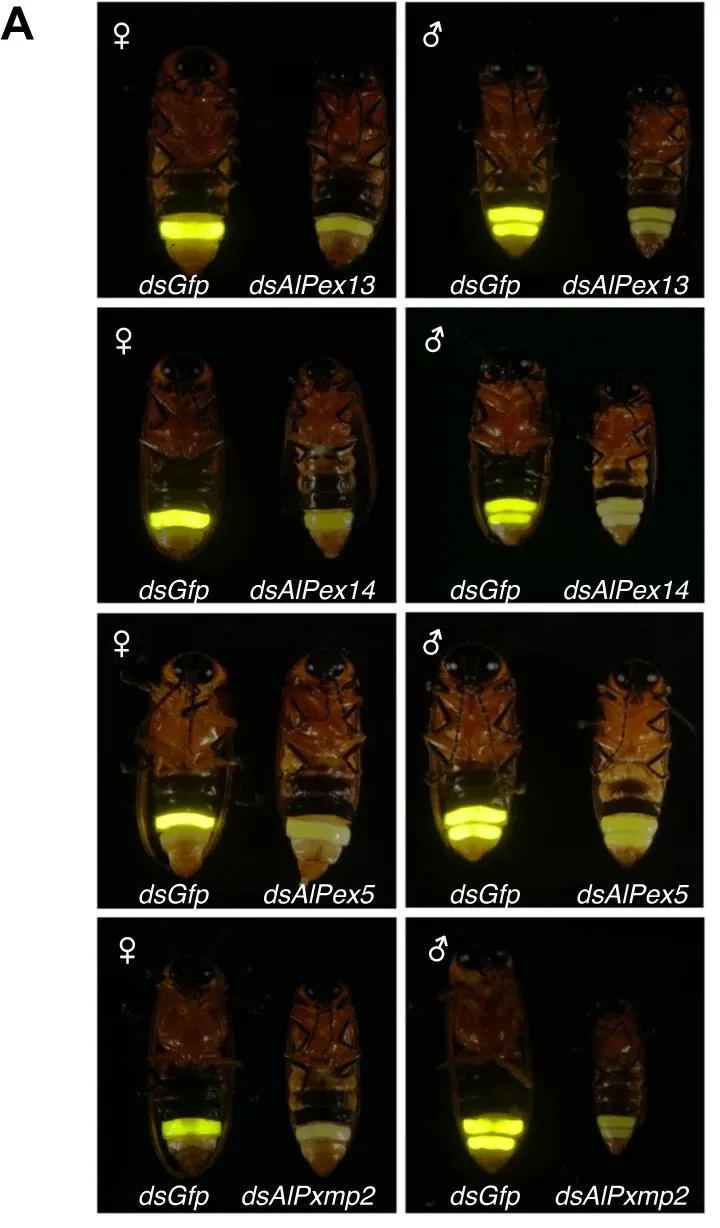
图6 参与AlLUC1转运至过氧化物酶体的pex基因的验证
总之,该研究揭示了两个关键的Homeobox转录因子调控萤火虫成虫发光器发育和生物发光的一系列新机制。AlABD-B 调控 AlUNC-4,它们相互作用。过氧化物酶 AlPex13 和 AlPex14 受到 AlABD-B 的调控。AlLuc1、AlPex5 和 AlPxmp2 同时受到 AlABD-B 和 AlUNC-4 的调控,但 AlABD-B 至关重要。AlUNC-4 提高了 AlABD-B 的转录活性,从而极大地激活下游基因的表达。AlPEX13 和 AlPEX14 相互作用,协助 AlPEX5 将 AlLUC1 运入过氧化物酶体。AlPXMP2 和 AlLUC1 相互作用,并参与将 AlLUC1 转运过氧化物酶体(图 7)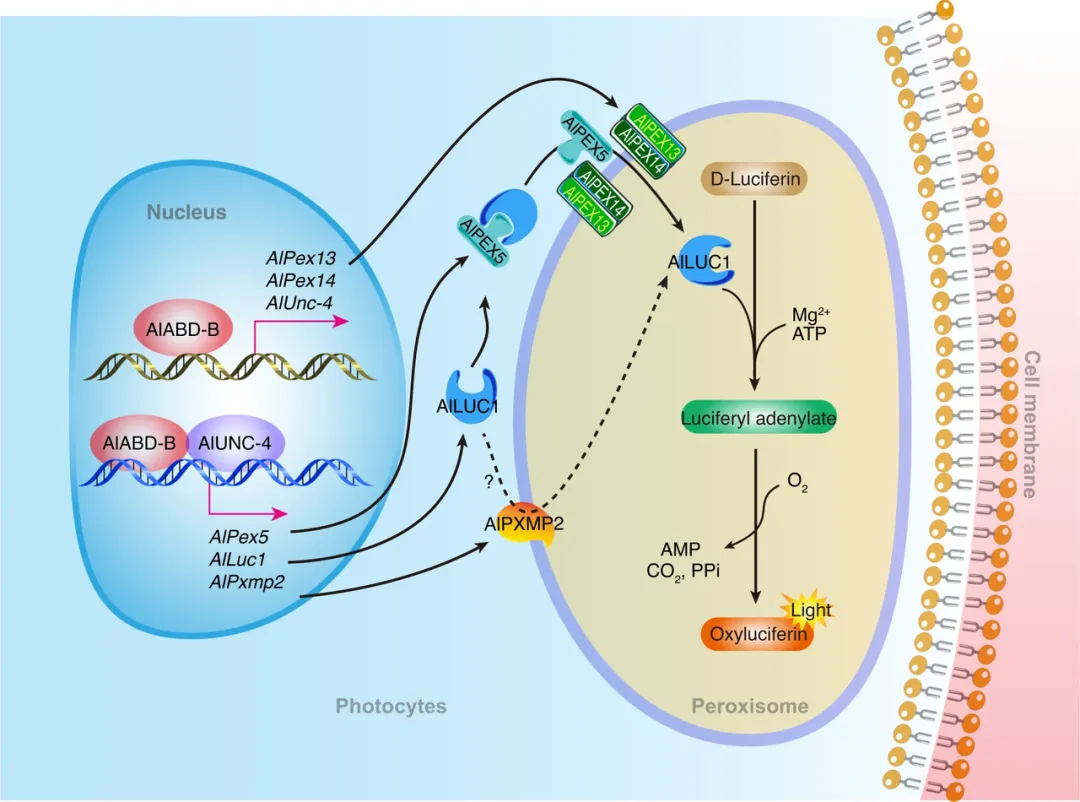










 张红雨教授参加开幕式并致辞
张红雨教授参加开幕式并致辞