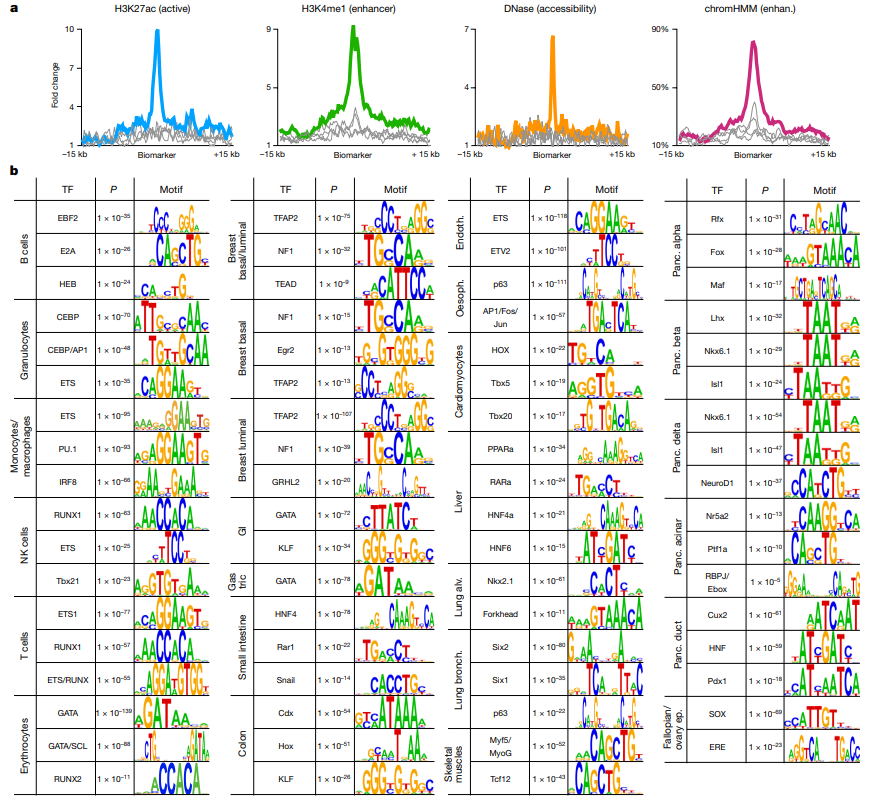
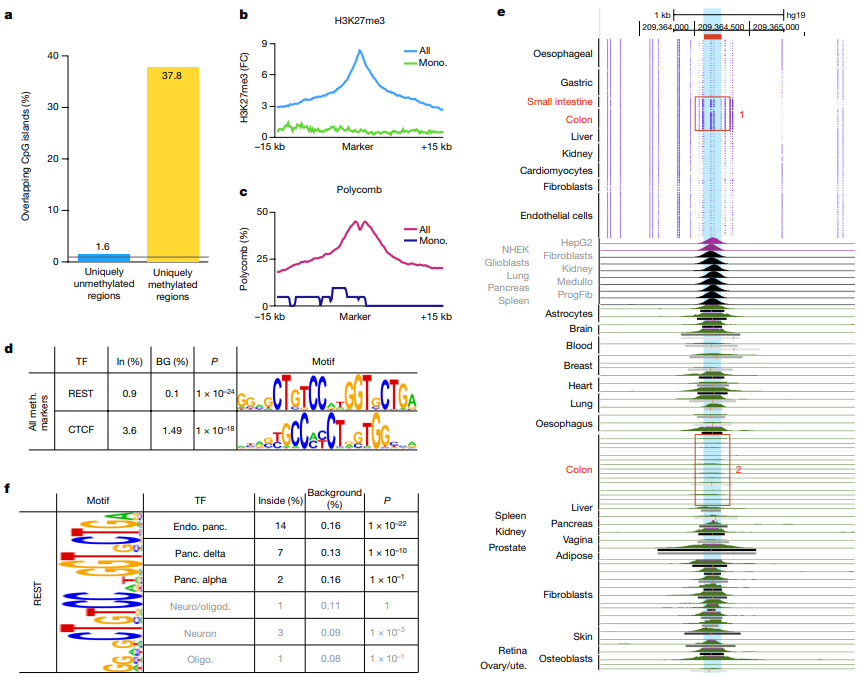
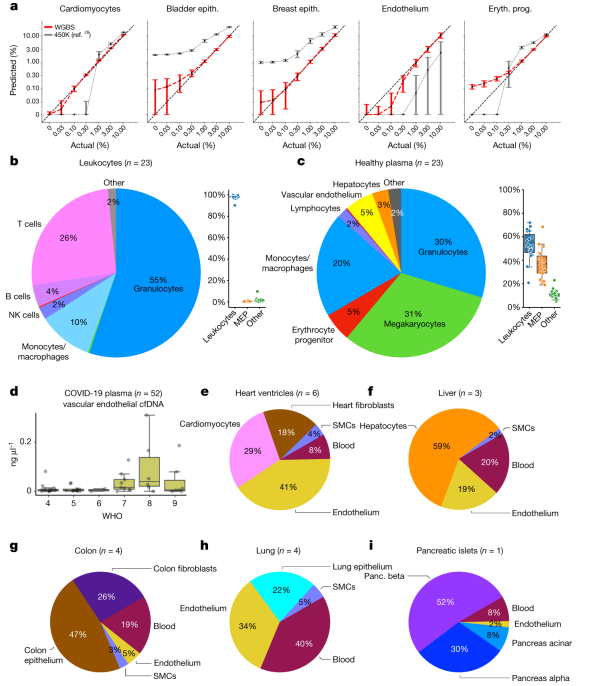
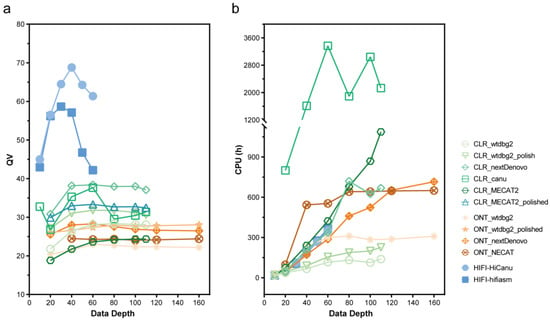
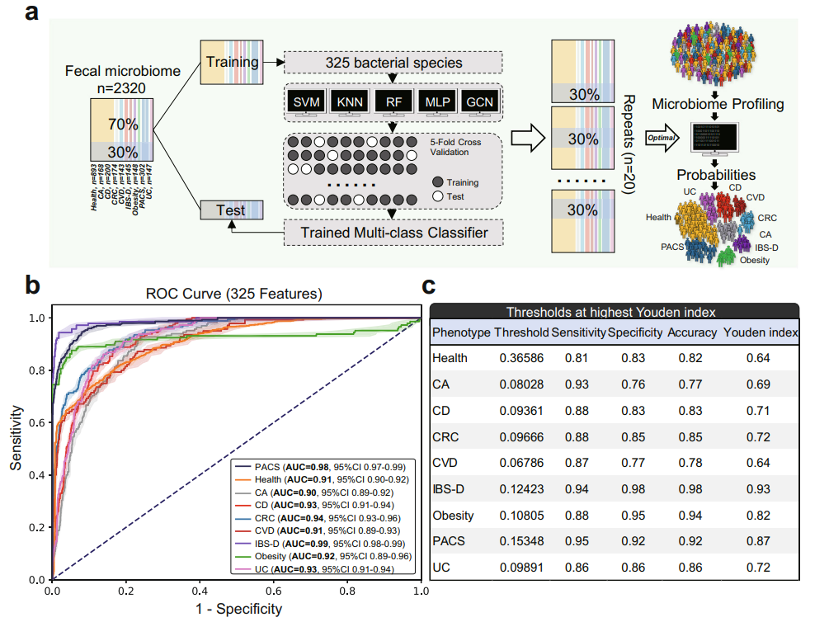
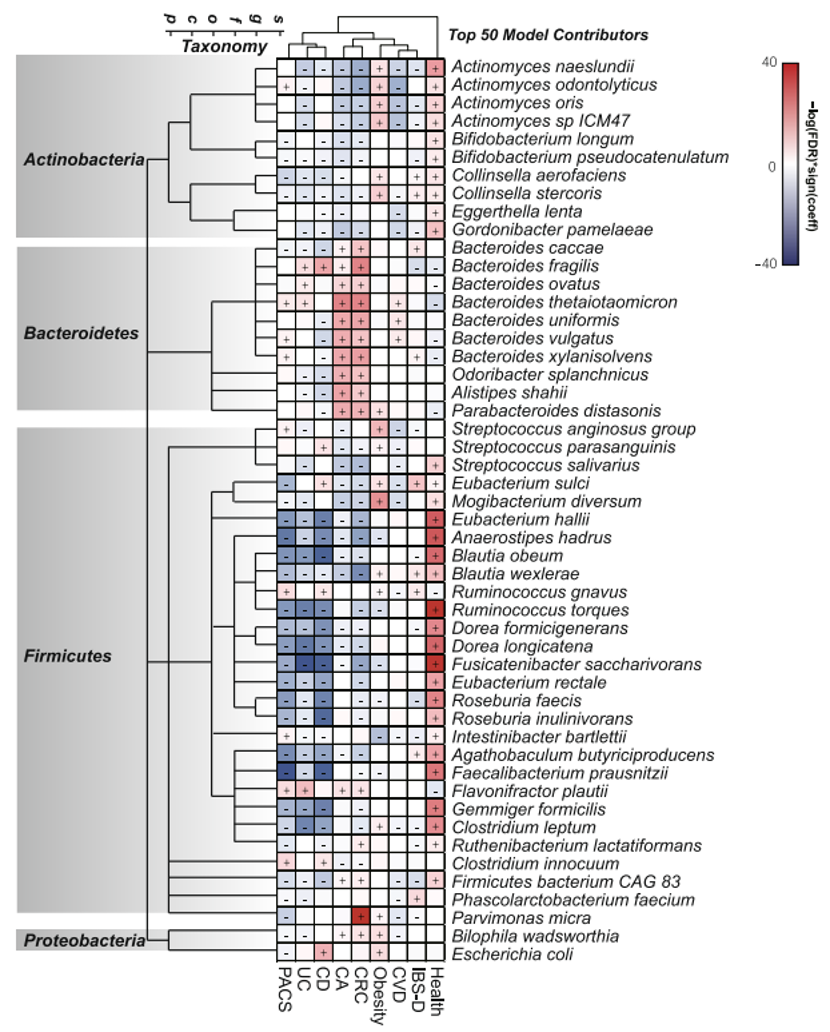
项目文章 | 长读长测序助力大麦基因组领域取得重要突破
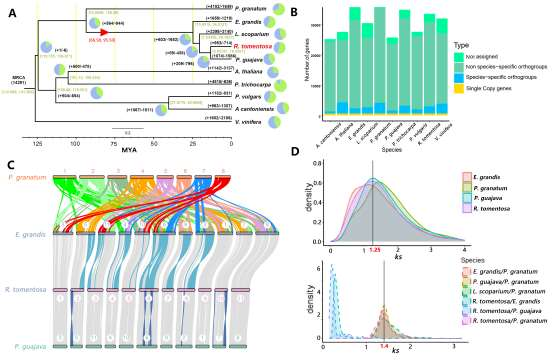
长江大学张文英教授课题组和澳大利亚技术科学与工程院院士、莫道克大学“西部作物遗传学联盟”主任李承道教授课题组合作,在植物科学国际权威杂志Plant Biotechnology Journal(影响因子13.263)在线发表题为“Genome architecture and diverged selection shaping pattern of genomic differentiation in wild barley”研究论文。研究者通过三代纳米孔测序,组装起源于以色列”进化峡谷”,峡谷南坡(干热的非洲坡)和北坡(冷湿的欧洲坡)的两个高质量野生大麦基因组。然后通过比较基因组学分析,群体遗传分析和转录组分析,研究了位于南坡和北坡两个野生大麦种群在非生物胁迫下的基因组分化和基因表达模式。
长江大学张文英教授,莫道克大学谈聪博士,莫道克大学胡海飞博士以及长江大学博士生潘锐为论文的共同第一作者,莫道克大学何田华博士,长江大学田小海教授和莫道克大学李承道教授为本文的共同通讯作者。本研究得到了中国国家自然科学基金,澳大利亚谷物研发公司(GRDC),主要粮食作物产业化湖北省协同创新中心、长江大学科技创新团队基金等项目的资助,并得到Claire Mérot, Peter Civáň 和Rajeev Varshney教授对文章的宝贵修改意见。希望组提供长读长测序服务与部分生物信息分析工作。

以色列”进化峡谷”是模拟研究非洲和欧洲不同生境生物进化与多样性的天然实验室。尽管地理接近,与北坡(冷湿的欧洲坡)相比,峡谷南坡(干热的非洲坡)的太阳辐射更高(多 200-800%),使得南北坡呈现出显著的环境差异和生物群体(动物、真菌和植物的种群)分歧。适应不同的环境可能会通过不同的选择压力,来驱动基因组区域的变化。通过比较种群的基因组差异,包括大的结构变异(SV)和单核苷酸多态性(SNP),可以揭示具有局部分化序列的基因组区域,并进一步研究其对物种环境适应性的影响。
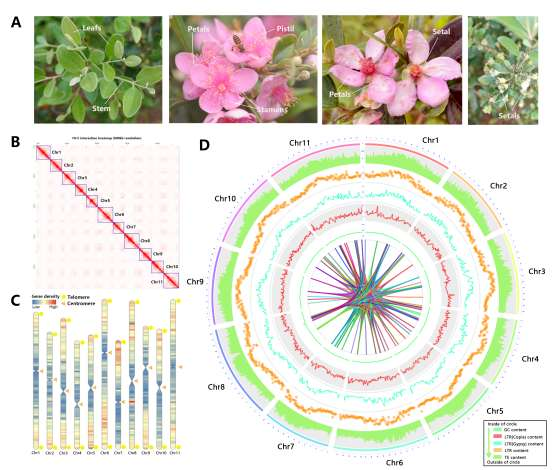
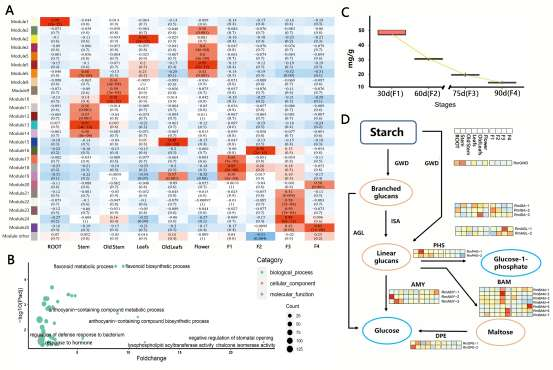
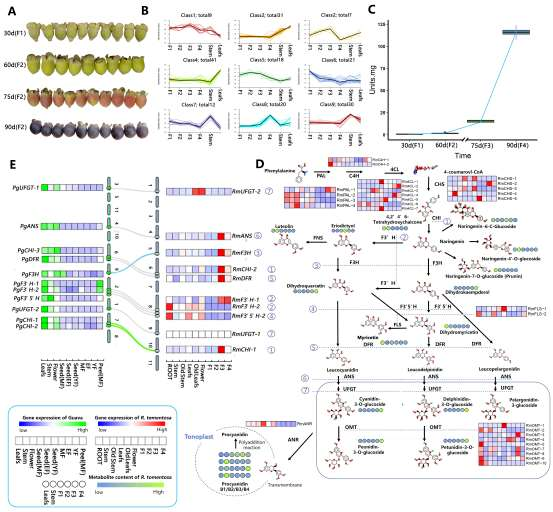
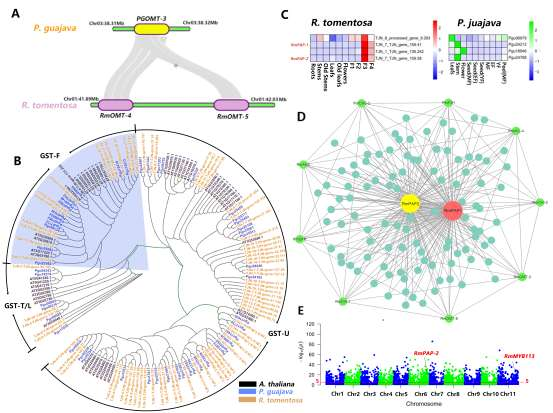
野生大麦(Hordeum spontaneum L.)是栽培大麦(Hordeum vulgare L.)的祖先,是栽培大麦改良重要的基因遗传资源。然而,我们对不同种群的野生大麦基因组分化和它们的基因组结构差异仍然知之甚少。在这里,研究者从头组装了分别位于以色列”进化峡谷”南坡和北坡(图1)的两个高质量野生大麦基因组 (图2)。位于南坡和北坡的两个野大麦种群具有共同的祖先,并且起源于地理上接近,但由于它们的不同的生长环境,导致经历了不同的选择压力。研究者进一步通过重测序和转录组手段研究两个种群在非生物胁迫下的基因组分化和基因表达模式。同时,发现了可能对分化表型产生重要影响的变异,例如影响两个野生大麦基因组之间的开花时间和干旱反应的变异。其中,一个29 bp 的启动子区域插入,在HvWRKY45基因中形成了顺式调控元件,这可能有助于增强南坡野生大麦对干旱的耐受性。启动子区域中的单个 SNP 突变可能会影响HvCO5表达并与其开花时间适应相关。研究者还揭示了两个群体之间的具有持续基因流动变,发现SNP 和小的 SV 通过局部适应与基因水平的遗传分化相关。相反,大的染色体倒位可能通过抑制染色体重组和基因流动形成染色体的基因组分化的异质模式(图3)。该研究为通过基因组研究环境适应性的遗传基础提供了新的见解,并为栽培大麦的遗传改良提供了宝贵的基因资源。

图1 以色列进化峡谷中的南坡和北坡的野生大麦种群,以及两个斜坡的气候差异
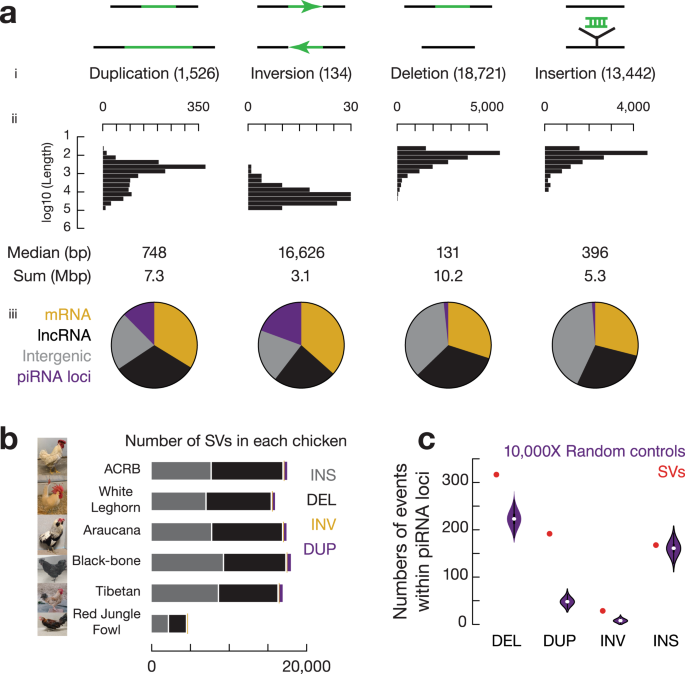
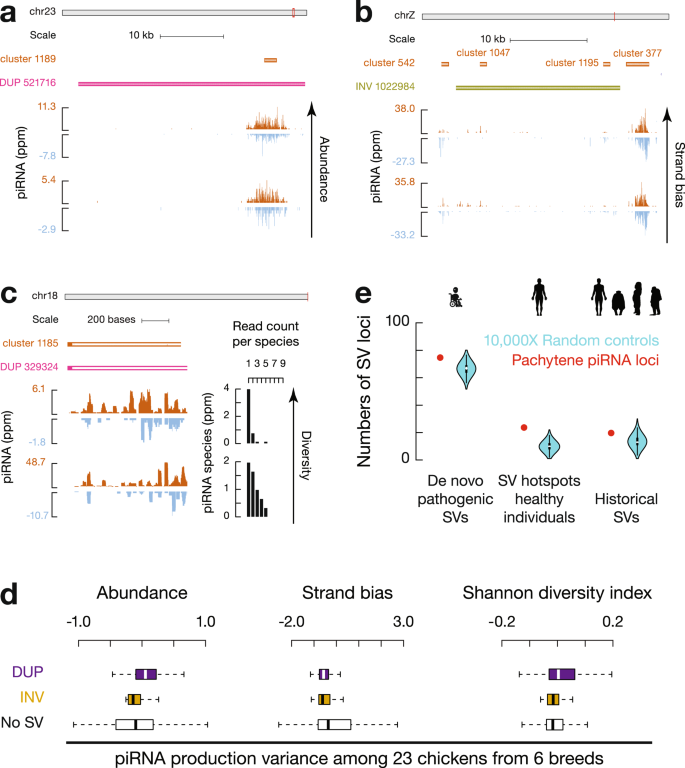
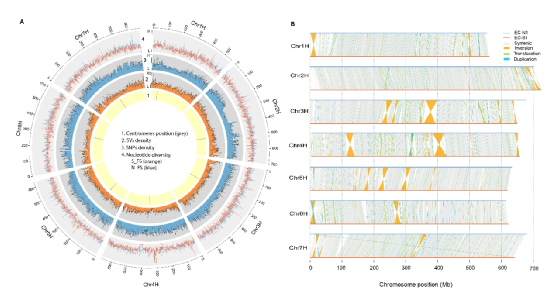 图2 野生大麦基因组组装与结构变异的新发现
图2 野生大麦基因组组装与结构变异的新发现
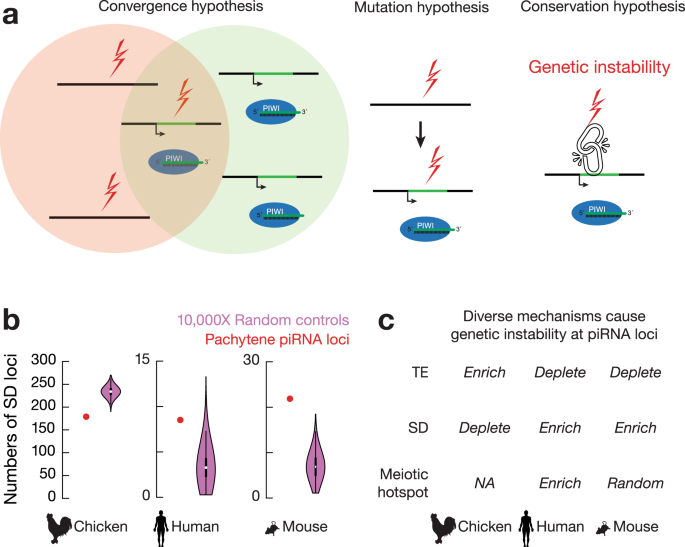
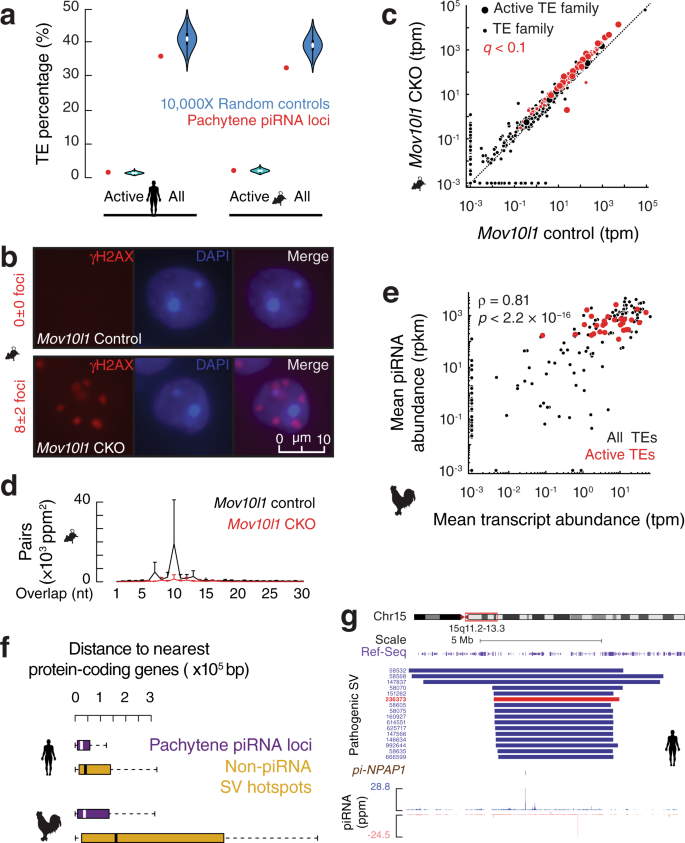
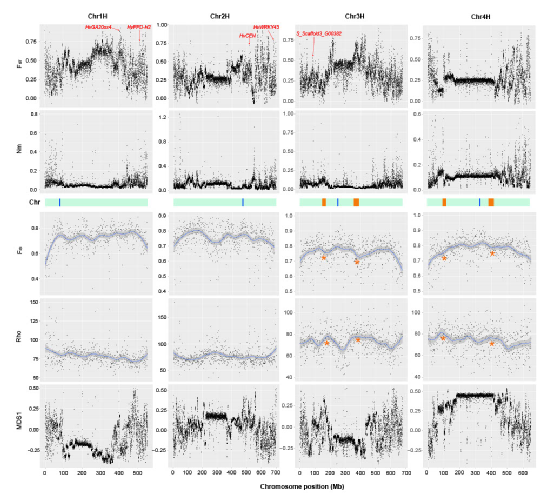 图3 大染色体结构变异对异质模式的影响
图3 大染色体结构变异对异质模式的影响



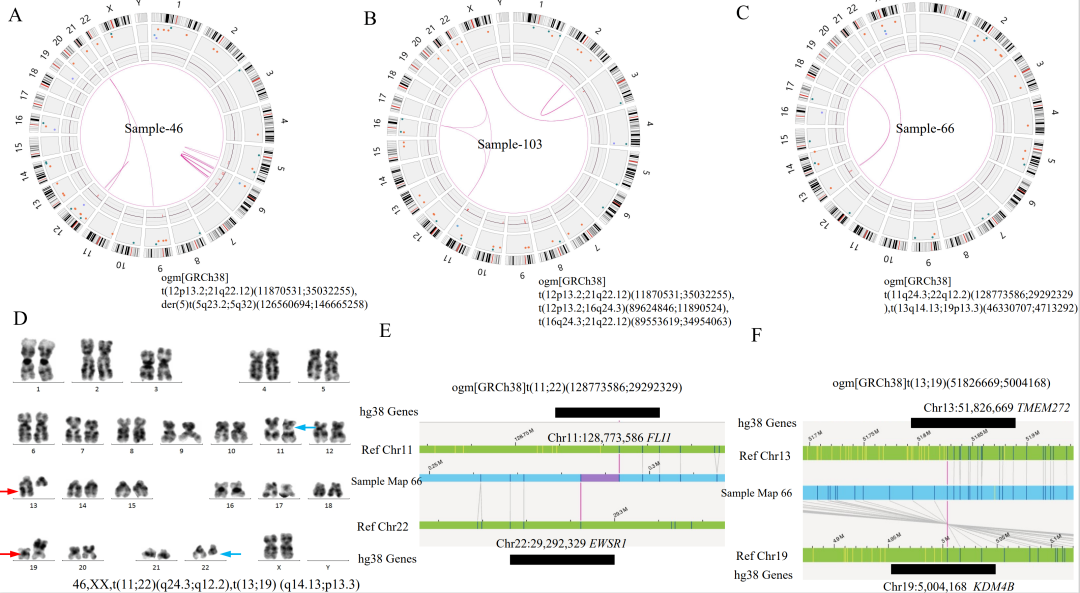
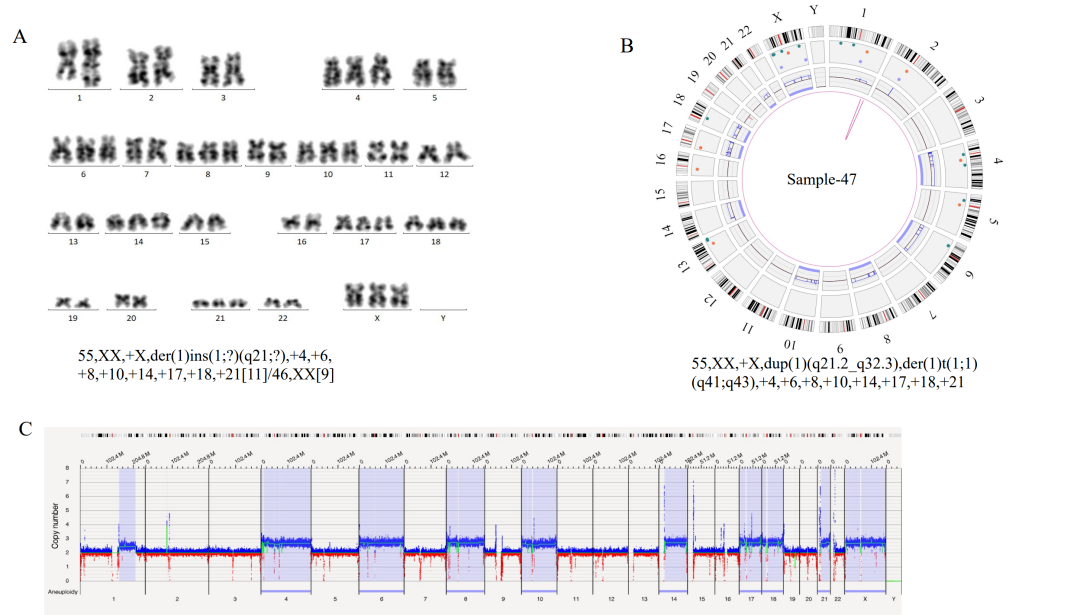
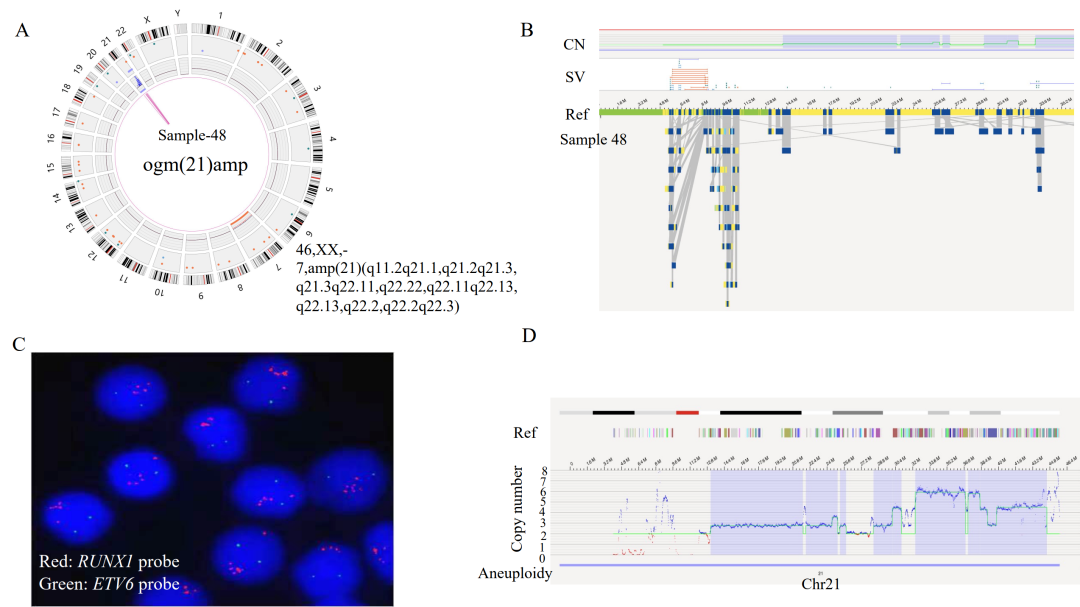
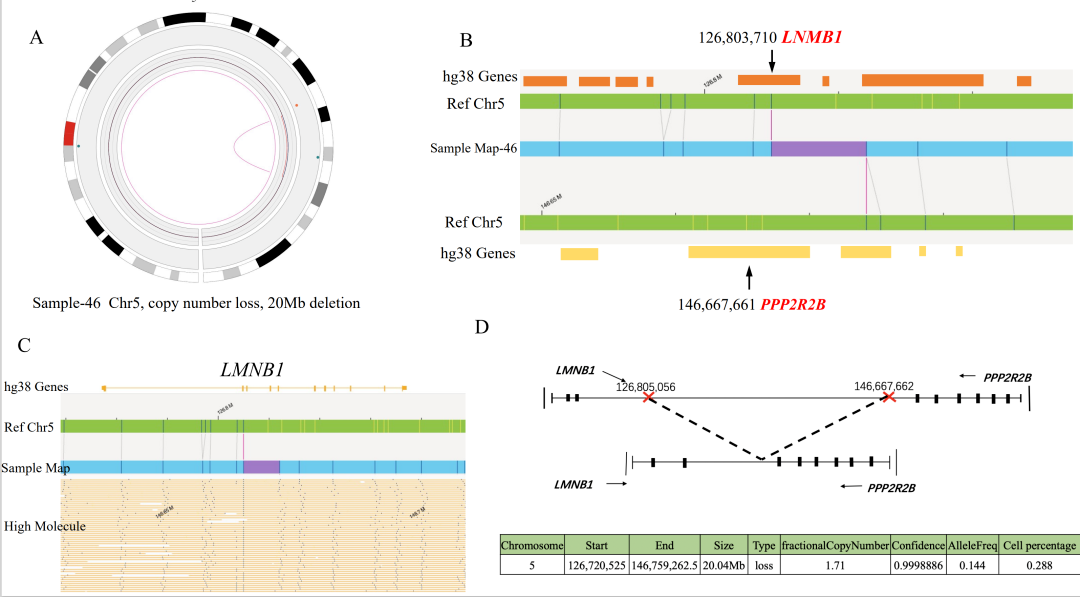
 注:在3个case中OGM未检出位于X染色体PAR区域的P2RY8::CRLF2融合,可能源于其VAF小于5%的检测下限,预计可在后续算法软件提升中解决该问题。
注:在3个case中OGM未检出位于X染色体PAR区域的P2RY8::CRLF2融合,可能源于其VAF小于5%的检测下限,预计可在后续算法软件提升中解决该问题。