项目文章||九江学院张化浩博士研究团队发表中国特有物种黑尾近红鲌高质量基因组


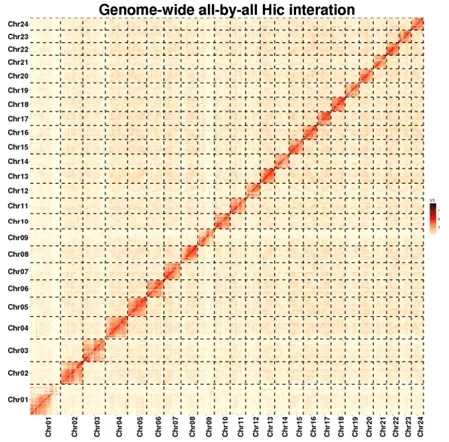
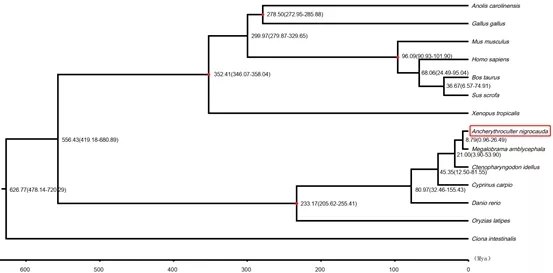
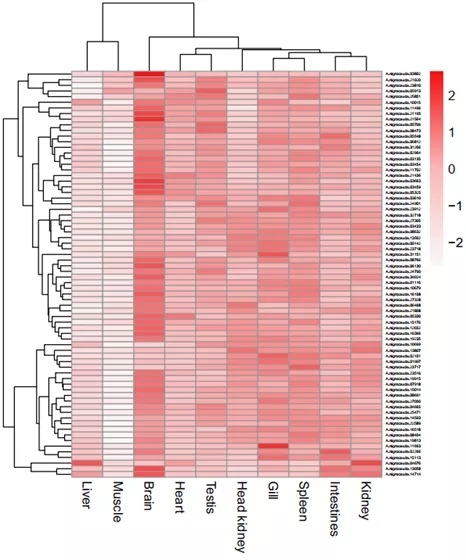

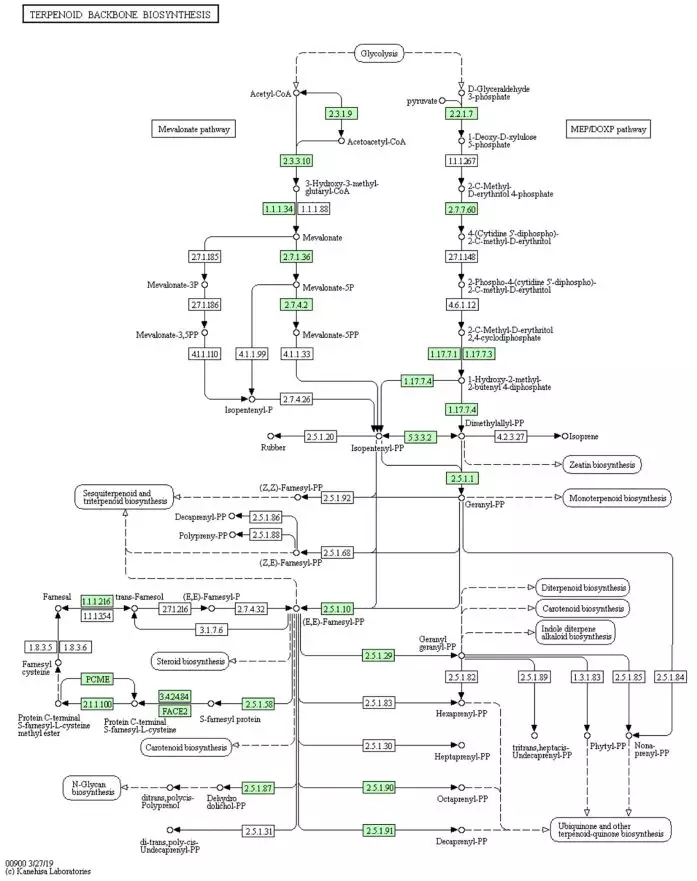
图1 萜类生物合成“KEGG通路图”,绿色方框基因在圆点斑芫菁基因组中发现。
蕨类在地球上已经存在了3.35亿年,是现存最古老的植物之一。远在恐龙出现和大陆漂移之前,它们便占据着原始大陆的沼泽森林,通过羽状叶片储存大量太阳能。蕨类死亡后的遗骸被埋葬在泥泞的沼泽沉积物中,经过千万年压缩转化成当代工业革命的能量——煤。

蕨类具有重要系统发育地位,尽管蕨类的基因组数据量仍然有限,但现有数据强烈表明,它们的基因组动力学与所有其他陆地植物截然不同。蕨类基因组的典型特征是染色体数目多,这被认为是通过多倍体的多个全基因组复制(WGD)周期产生的。然而,与被子植物多倍体相比,蕨类的多倍体后二倍体化过程通常涉及基因沉默而不是DNA消除,从而导致染色体数目异常增多,同时保持二倍体基因的表达[1]。蕨类染色体的平均数目(n = 63.5)[2]是被子植物平均数目的三倍多(n = 21.55)[3]。而蕨类基因组大小平均为12Gb[3],最大甚至达到148Gb[4,5]
 蕨类基因组研究现状
蕨类基因组研究现状




表2 NextDenovo的组装案例

2019年11月1日,北京市农林科学院许勇团队、中国农科院郑州果树所刘文革团队、美国康奈尔大学Boyce Thompson研究所费章君团队和中国农科院深圳基因组所黄三文团队等合作在国际学术期刊Nature Genetics在线发表了题为Resequencing of 414 cultivated and wild watermelon accessions identifies selection for fruit quality traits的研究成果。该研究利用三代测序技术完成了西瓜品种“97103”新的基因组精细图谱绘制,结合414份西瓜二代重测序数据,利用群体基因组分析及全基因组关联分析对西瓜的进化、驯化历史进行了解析。武汉未来组承担了该研究中的PacBio基因组测序、HiC测序、Bionano测序以及PacBio全长转录组测序工作。

西瓜(Citrullus lanatus, 2n=2x=22)是全世界最普遍的水果之一。它起源于非洲,隶属于葫芦科西瓜属,其驯化历史已超过4000年[1]。在漫长的驯化过程中,自然选择和人类选择是如何导致西瓜的表型发生显著性改变,目前还未完全清楚。




群体遗传学研究的一个重要手段是利用高通量测序技术提供的DNA序列变异信息来推测作用于基因组的各种力量(突变,自然选择,群体结构,杂交等)是如何影响生物演化进程的。目前进行DNA序列变异分析的主要策略包括:基于比对(Alignment)检测和基于组装(De novo)的方法。基于序列比对的群体重测序凭借其高性价比,是动植物分子育种、群体进化研究中最为迅速有效的方法之一。但是随着测序成本的降低以及群体研究的深入,基于组装的群体基因组De novo越来越多的应用于群体遗传学研究,尤其是在解决物种进化的重大问题上表现抢眼。

图1 基因组结构和基因渗入形成了蝴蝶多样性
袖蝶属是由至少40个蝴蝶品种组成的多样化属,它们以其多彩且极为独特的翅翼图案而闻名。由于这种多样性,该物种一直被用于研究物种之间的基因渗入。然而,由于难以区分基因混合,杂交在袖蝶属适应性辐射中的作用仍然受到质疑。本研究构建了20个新的袖蝶属蝴蝶从头组装基因组,并使用一种新方法确认通过杂交引入的基因变异,结果表明基因组结构和基因渗入形成了蝴蝶多样性,物种间偶尔的基因渗入和重组可能会对基因组产生重大的长期影响,并为物种的快速适应性分化和辐射提供必要的基因物质。
 图2 大规模的反刍动物基因组测序为研究反刍动物的进化和特性提供了新的思路基于三代测序的群体基因组De novo研究也如火如荼,美国约翰霍普金斯大学、冷泉港实验室和其他机构的研究人员使用Oxford Nanopore长读长技术的高通量PromethION测序平台,在100天内对100个番茄品种完成测序。研究者结合使用长读长技术、计算生物学和功能研究来发掘和鉴定番茄的结构变异,以便在未来进行从自然变异和驯化到作物改良的一系列研究。该团队同时采用了比对检测和从头组装两种策略进行结构变异分析,2019年5月7日,Nature Plants杂志在线发表了该研究的部分成果[3]。
图2 大规模的反刍动物基因组测序为研究反刍动物的进化和特性提供了新的思路基于三代测序的群体基因组De novo研究也如火如荼,美国约翰霍普金斯大学、冷泉港实验室和其他机构的研究人员使用Oxford Nanopore长读长技术的高通量PromethION测序平台,在100天内对100个番茄品种完成测序。研究者结合使用长读长技术、计算生物学和功能研究来发掘和鉴定番茄的结构变异,以便在未来进行从自然变异和驯化到作物改良的一系列研究。该团队同时采用了比对检测和从头组装两种策略进行结构变异分析,2019年5月7日,Nature Plants杂志在线发表了该研究的部分成果[3]。以上案例表明随着群体遗传学研究的深入,基于群体基因组De novo的群体基因组学研究是大势所趋,希望组科技服务隆重推出群体基因组De novo服务:
希望组3大实力保证交付质量与周期:
测序产能——ONT P48 产能怪兽


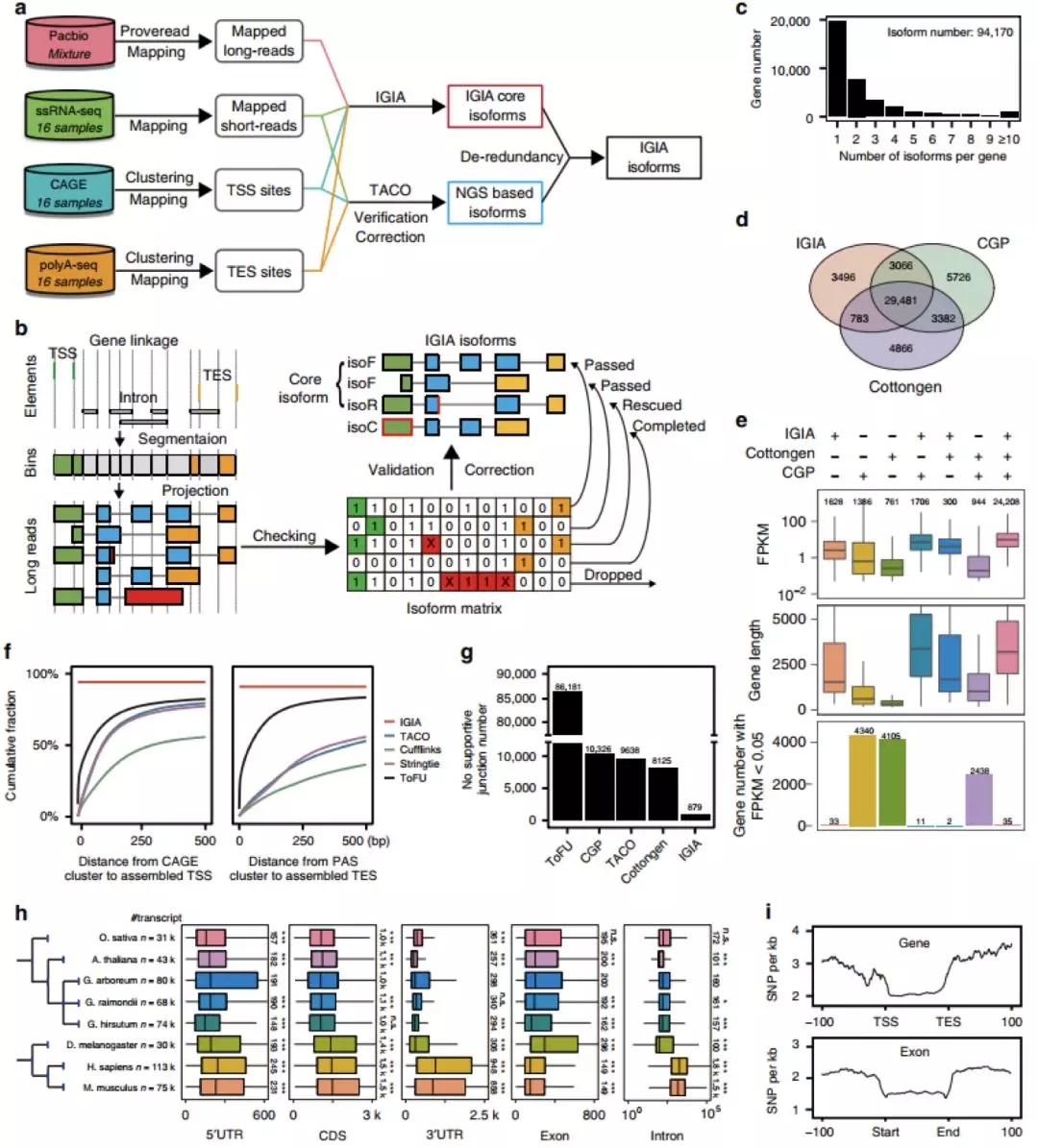
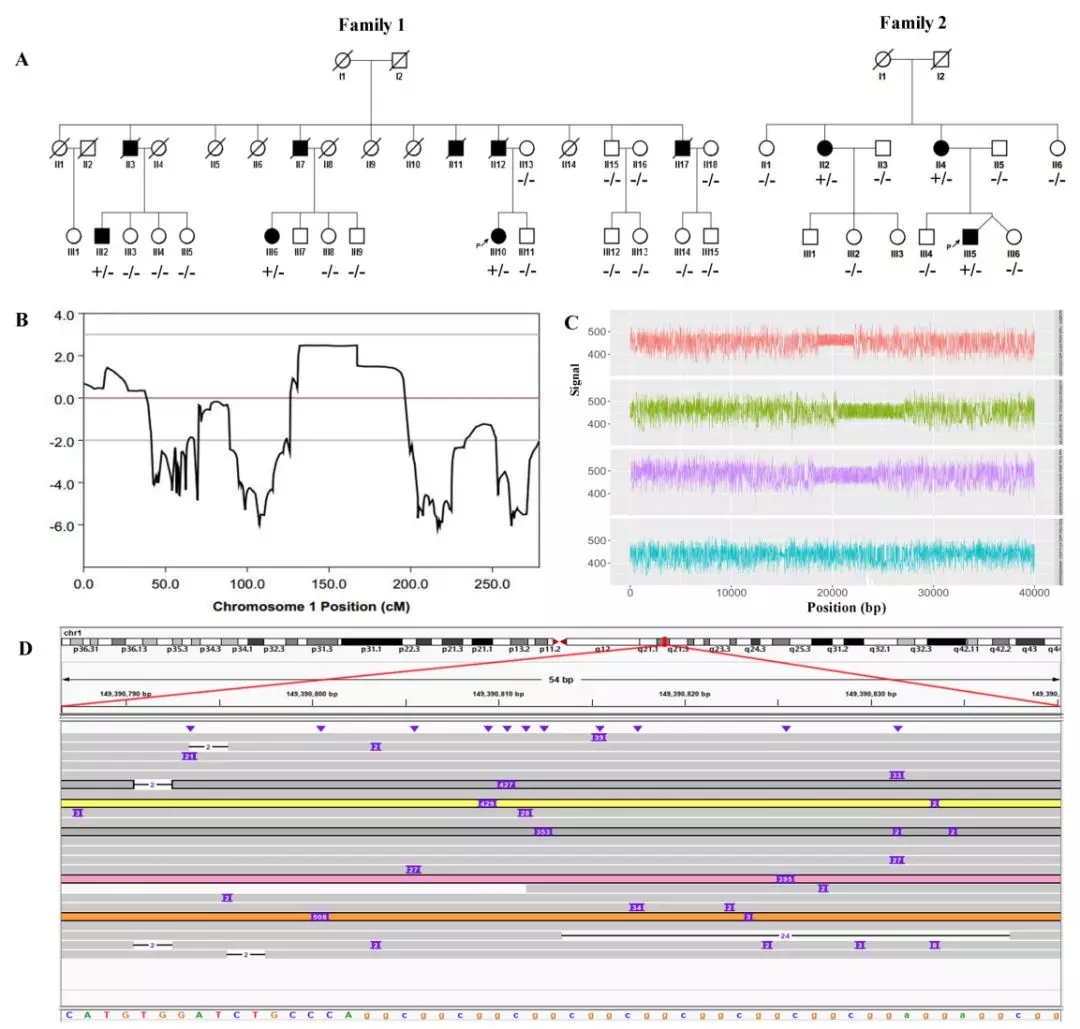
图1 针对亚洲棉高分辨率转录组景观设计的多重RNA-Seq策略。
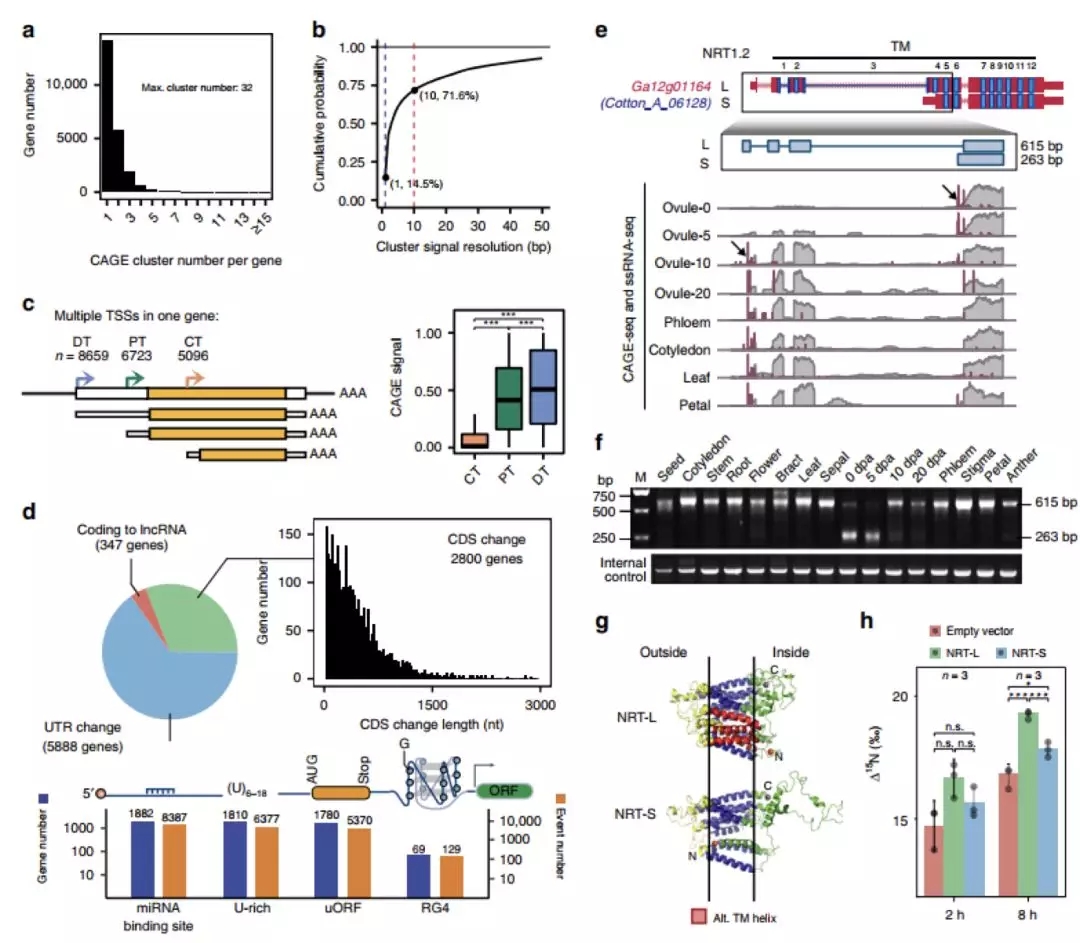
图2 亚洲棉中多转录起始位点和可变启动子的使用。
于PolyA-seq的3’末端信息,我们对16个组织中所有表达基因的全基因组TES进行了分析。揭示了它们的序列特征、发育过程中的动态规律和组织规范。结果表明在棉花中所有表达的基因中,有40.2%的人至少有两个TES,基因的3’末端同样存在多TES调控的现象,在发育和组织分化过程中,很多基因的转录终止通过可变的TES调节其3’UTR的长度。
本研究基于IGIA注释,对亚洲棉的23,451个多外显子基因进行了可变剪接(AS)的系统分析。结果表明,所有AS事件中内含子保留(RI)占62.2%(图3a),在所有报道的植物中是最高的。此外,某些基因中的几个区域显示出高度丰富的AS事件,研究者将其称为AS热点。进一步分析表明,大多数AS热点影响保守的蛋白质结构域(图3g-h)。微外显子(micro-exon)是动物中首先报道的一种微小外显子,其长度短至仅3nt,该研究通过系统分析,首次在棉花中鉴定到微外显子的存在,还通过在多个植物物种的比对,发现了一个具有潜在重要作用的45nt的保守微外显子。
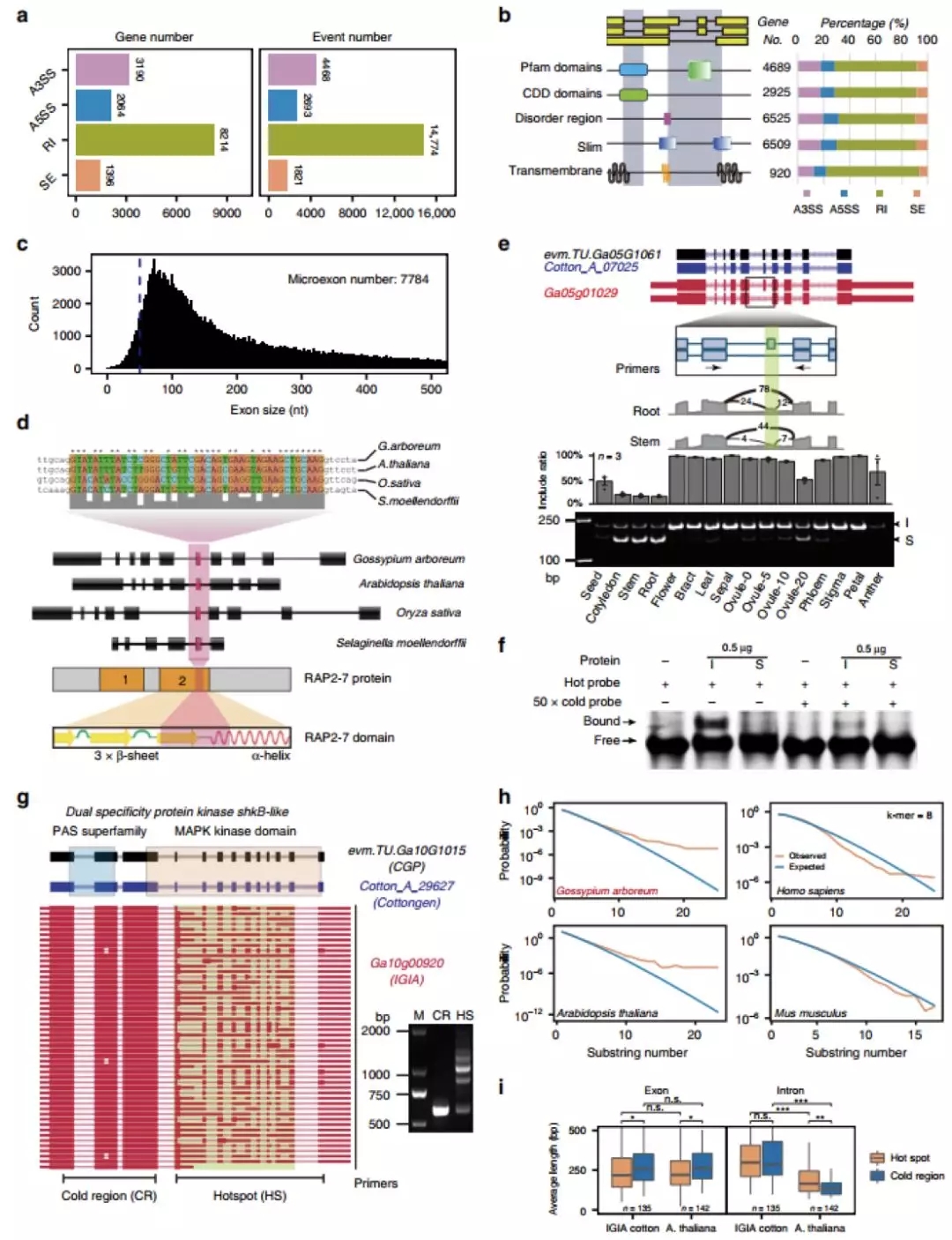
图3 亚洲棉中选择性剪接调控及热点
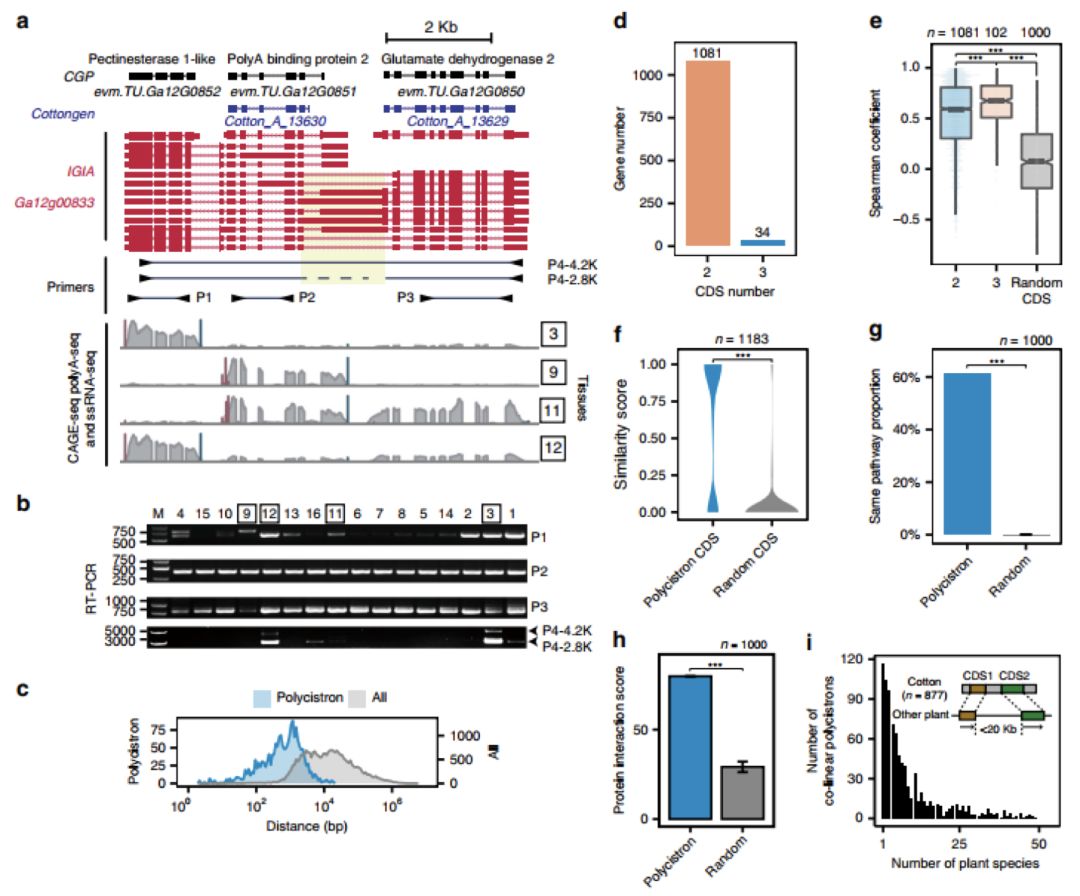
本研究通过IGIA算法整合了四种互补的高通量技术:用于直接读取全长转录本异构体的PacbioIso-seq,用于定量表达和剪接的链特异性RNA-seq,准确定义转录起始和终止位点的CAGE-seq和检测聚腺苷酸化位点的PolyA-seq,生成了高分辨率的亚洲棉转录组景观图谱。发现并验证了棉花发育中基因表达调控的不同模式,如可变启动子和终止子调节、微外显子剪接、多顺反子转录通读和RNA选择性剪接热区等复杂现象,对未来棉花功能基因组学的进一步发展意义重大。
参考文献:
Wang K, Wang D, Zheng X, et al. Multi-strategic RNA-seqanalysis reveals a high-resolution transcriptional landscape in cotton[J].Nature communications, 2019, 10(1): 1-15.
继8月底宣布单Cell reads N50突破100Kb后,希望组ONT Ultra-long测序再传捷报,某单子叶植物超长测序数据产出超过1200Gb,平均读长N50达51.9Kb,单cell最高产出58.9Gb,最长reads N50达143.3Kb!这标志着希望组ONT Ultra-long测序已经能够稳定产出高质量的超长片段!

某单子叶植物ONT Ultra-long测序单Cell产量超过10G,Reads N50达143.3Kb,长度100Kb以上的reads占总数据量的65.3%,长度200Kb以上的reads占总数据量的28.1%!

图2 某单子叶植物单Cell 超长测序读长分布
希望组自2017年推出ONT超长测序服务以来,现已完成昆虫、两栖动物、鱼类、鸟类、哺乳动物、多倍体植物、药用植物等数百个物种的ONT Ultra-long测序工作,并且多个物种测序单Cell read N50突破100Kb!

牛津纳米孔测序平台独有的Ultra-long测序能够产生超长测序片段,轻松跨越基因组中连续重复或大片段重复区域,更大限度地还原真实的基因组景观。
对于基因组中“暗区”,二代测序小短腿直接掉入深渊,三代测序小心翼翼能够跨过,而Ultra-Long Reads能够轻松跨越连续重复区域,提供更多的序列信息,更便于组装过程重复片段划分。
在基因组组装过程中可以通过增加读长获得理想组装质量[1],加入Ultra-Long Reads数据可以显著提升人类基因组组装效果,填补基因组中的缺口,甚至组装出端粒到端粒水平的完整染色体[2]。
相同测序深度下采用Ultra-Long的建库测序方法,产生用于组装超大型基因组的read数更少,降低了组装复杂度,减少了计算资源的使用,能够节省一定的组装成本。
Nanopore Ultra-long 超长读长的 Reads N50 相比 Normal long 有成倍的提升,在基因组组装过程中加入适量 Ultra-long 数据,可有效提升基因组组装质量。高杂合、高重复基因组采用纯 Ultra-long 数据进行基因组组装,能够达到较好的组装质量。
希望组ONT Ultra-Long组装案例


希望组三代测序组装采用PromethION 48+Ultra-long+Next系列组装软件+Bionano&Hi-C的最新策略,结合华为云将纳米孔测序数据分析流程整合到云计算平台上,实现急速基因组组装与注释,为全球客户提供快速、高效的纳米孔长读长测序计算和存储服务!希望组三代测序,技术顶尖,算法领先,服务全面,为您的科研之路保驾护航!
[1] Henson J, Tischler G, Ning Z. Next-generationsequencing and large genome assemblies[J]. Pharmacogenomics, 2012, 13(8):901-915.
[2] Jain M, Koren S, Miga K H, et al. Nanoporesequencing and assembly of a human genome with ultra-long reads[J]. NatureBiotechnology, 2018, 36(4).
结构变异(Structural variants,SVs)通常是指基因组上大长度的序列变化和位置关系变化。研究表明,与单核苷酸多态性(SNPs)相比,SVs可以解释更多的表型变异。在植物基因组中,SVs的类型、大小以及对于表型的贡献多有报道,大概1/3已报道的作物表型是由于结构变异引起的(Gaut et al. 2018 Nature Plants),但是对于SVs在种群个体间的分布以及种群动态,人们知之甚少。希望组科技服务在6月份推出了基于Nanopore平台的三代测序群体基因组SVs研究,许多老师对这一研究非常感兴趣,但苦于没有研究思路。最近,Nature Plants杂志在线发表了一篇群体水平结构变异研究文章,加州大学Irvine分校周永锋博士为第一作者,Brandon Gaut教授(UC Irvine)和Dario Cantu教授(UC Davis)为共同通讯作者。该研究探讨了葡萄驯化过程中结构变异的群体遗传学,今天就给大家分享一下这篇文章的研究策略,给各位提供一些科研灵感。
研究背景
多年生植物栽培葡萄(Grapevine)是由其野生祖先欧亚葡萄(Eurasian grapevine),在约8000年前的高加索地区驯化而来。驯化提高了果实含糖量,增大了果实的体积和串大小,改变了种子形态,同时使雌雄异株转变为雌雄同体无性繁殖。无性繁殖作物处于永久性杂合状态,并随着时间累积体细胞突变(Zhou et al. 2017 PNAS)。理论上,雌雄同体葡萄可以自交,但实践中,其自交后代无法存活,可能是近亲繁殖暴露了杂合状态下的有害等位基因。因此,大多数葡萄品种是远源亲本之间的杂交种,加上体细胞突变的积累,导致葡萄品种往往是高度杂合的。本研究通过调查野生和驯化葡萄中SV的群体遗传来填补我们对植物基因组进化认知的空白。
研究策略

无性系繁殖葡萄基因组中肆虐的半合子状态
研究者首先利用三代测序+二代测序+Hi-C技术,组装了高杂合葡萄霞多丽品种的基因组序列,并对其进行了注释和评估,发现无性系繁殖葡萄基因组中有七分之一(~15%)的基因属于半合子,这一结果在黑比诺(PN40024)基因组与赤霞珠(Cab08)参考基因中得到了验证。
随后研究者用长、短reads比对和全基因组比对等方法,综合比较了Char04和Cab08两基因组之间的SVs。结果表明利用长reads比对检测到59,913个SVs,其中75%得到另外两种方法的证实。两个品种之间有近5%的PAV基因差异,半合子基因差异高达25%,表明葡萄品种之间显著的结构变异(图2)。

图2 高杂合Char04及与Cab08结构变异的比较
SVs群体遗传分析
为了获得更广泛的葡萄品种及其野生亲缘SVs信息,研究者收集了有代表性的50个栽培葡萄品种和19个野生亲缘品种的短读长测序数据。以Char04为参考基因组,以Char04和Cab08综合比对的交叉SVs集合为金标准,获得了一组高度筛选的481,096个SVs。
随后,研究者利用上述SVs集合计算了12个野生种和12个栽培种的SFS(图3),推断了对SVs类型的选择强度,并对比了驯化和野生祖先之间的SVs频率。结果非同义SNP(nSNP)和SVs都经历了强烈的纯化选择,不同SVs类型中,易位TRAs和倒位INVs的选择性更强。因此SVs事件比nSNP更有害,INV和TRA事件尤其有害。

图3 处于强烈净化选择中的有害SVs
SVs在无性系繁殖体中积累
研基于SNP的个体杂合度分析,栽培葡萄个体杂合度要高出野生型葡萄11%,相应的单个栽培种SVs比野生种高出6%,但纯合子SVs或推测为中性的sSNPs没有明显增加(图4a)。因此有害SVs在无性繁殖情况下以杂合隐性形式隐藏、积累。
杂合变异的积累会影响连锁不平衡(LD),通过测量SVs、SNP和组合数据集的LD随物理距离的下降来分析SVs的种群频率。结果发现,与野生品种相比,栽培品种的LD下降速度更快;与SNP相比,SVs的LD下降更快;下降速度最快的是SV+SNP数据集。表明由于有害影响,SVs通常比SNP的种群频率更低。


图4 葡萄驯化相关SVs的群体遗传学
大的,独立的倒位对浆果颜色的影响
通过计算固定指数(FST)来估计SNP和SVs在基因组中的差异(图4c),在2号染色体上发现了两个异常值分别与性别决定和浆果颜色相关。其中对浆果颜色相关区域的进一步研究发现,在霞多丽中有一个4.82Mb的倒位(图5b),并有证据支持白浆果的独立起源通常是由这种倒位介导的(图5d),其导致了半合子状态的花青素合成基因MybA1和MybA2的等位基因空缺。

图5 与白色浆果相关的染色体倒位
小结
本研究首先组装了高杂合葡萄霞多丽的基因组序列,评估了该基因组中SVs类型和分布以及导致遗传半合子的SVs。随后将霞多丽与赤霞珠基因组进行综合比较,获得了一套种间SVs标准集,并以此指导、推断栽培葡萄及其野生祖先群体样本中的SVs。然后利用获得的群体SVs数据集,推断不同类型变异的选择强度,探讨了在栽培葡萄上由异交向无性繁殖转变的效应,最后研究了栽培葡萄与其野生祖先之间SVs差异特别显著的与浆果颜色相关的基因区域。 在结构变异的研究中,最首要的任务是获得到研究对象全面、准确的SVs集合,本研究中作者为了获取准确的SVs集合,利用三代测序组装了霞多丽基因组,采用长读长比对来鉴定SVs,基因组比对和短读长比对进行验证,短读长仅检测到长读长比对检测数量的62%,长读长检测的SVs中75%得到另外两种方法的验证。可见相比短读长利用三代测序检测的SVs更加全面准确。
参考文献:
Gaut B S, Seymour D K, Liu Q, et al. Demography and its effects on genomic variation in crop domestication[J]. Nature plants, 2018, 4(8): 512.
Zhou Y, Massonnet M, Sanjak J S, et al. Evolutionary genomics of grape (Vitis vinifera ssp. vinifera) domestication[J]. Proceedings of the National Academy of Sciences, 2017, 114(44): 11715-11720.

希望组

希望组科技服务

希望组诊断服务